
原子吸附的二维CrI3铁磁半导体电学和磁学性质的第一性原理研究*
2021-06-18 08:41秦文静徐波孙宝珍刘刚
物理学报 2021年11期
秦文静 徐波† 孙宝珍 刘刚
1) (江西师范大学物理与通信电子学院, 计算材料物理实验室, 南昌 330022)
2) (江西师范大学物理与通信电子学院, 凝聚态研究所, 南昌 330022)
采用第一性原理的方法系统研究了碱(土)金属原子、过渡金属原子和非金属原子吸附的二维CrI3单层的电学和磁学性质.结果表明, 金属原子倾向于吸附在6个I原子形成的环中心并且与Cr原子处于同一高度, 非金属原子吸附在I原子形成的环内, 但位置则依赖于原子种类.除Ti和Mn原子之外的其他原子的吸附并未改变CrI3单层的本征铁磁半导体属性, 而Ti和Mn原子的吸附则使CrI3单层转变为反铁磁构型.金属原子的吸附导致Cr原子局域磁矩增加, 但不超过4μB; 而非金属原子的吸附则使Cr原子的局域磁矩呈现多样化.结合分波态密度我们对Cr局域磁矩的变化机制进行了细致分析, 发现Cr磁矩的增加与电荷转移直接相关.我们的研究结果为铁磁半导体CrI3的性能调控提供了新的思路, 在未来自旋电子学中有着潜在的应用前景.
1 引 言
自石墨烯[1-4]被发现以来, 原子层厚度的二维材料一直备受人们关注.近年来, 过渡金属硫族化物及其衍生物[5-10], 六方氮化硼[11]和黑磷[12,13]等也加入了二维材料家族, 在光电子器件[14], 自旋电子器件[13,15-17], 储能器件[18-20]和生物传感器等领域有着广泛的应用.由于难以寻找具有本征铁磁性的二维材料, 因此二维材料在自旋电子学中的应用受到了限制.最近, 单层三碘化铬(CrI3)[21]在实验上的成功合成引起了人们的关注.CrI3是一种具有本征铁磁性的半导体材料, 其居里温度为45 K[21],且有合适的带隙(带隙约为1.53 eV)[22], 因此是二维铁磁性半导体研究领域的重要突破.通常情况下, 铁磁性的材料具有金属性, 而反铁磁性的材料具有半导体特性.因此, 获得铁磁绝缘体或反铁磁金属更能引起人们的关注.
自二维CrI3单层问世以来, 研究者们对其电、磁学性质[22-26], 光学性质[27,28], 拓扑绝缘性质[29,30]等方面进行了相关研究, 其中涉及CrI3本征铁磁性的研究居多.不仅如此, 人们还通过应变[31-33],载流子掺杂[34,35], 缺陷引入[36-38], 施加电场[39,40]和表面吸附[41-44]等方法对CrI3单层的性质予以调控.在已有的研究中, Zheng等[45]对CrI3单层施以应变, 发现压缩应变可以导致CrI3从铁磁半导体到反铁磁半导体的转变, 而拉伸应变则能将磁序从平面外翻转到平面内.Gao等[46]通过引入Cr缺陷使CrI3半导体半金属化, 且半金属化的状态与Cr缺陷的浓度无关.我们之前的工作[47]也证实了I缺陷能够增强CrI3单层的磁性, 同时载流子掺杂可以进一步调控磁性.施加电场[48]对CrI3单层的结构和磁性质会产生巨大影响, 可以实现铁磁构型到反铁磁构型的转变.Guo等[49]在CrI3单层表面吸附Li原子, 使CrI3单层半金属化,同时提高磁矩和居里温度.
对于层状磁性材料而言, 在众多的性质调控手段当中, 表面原子吸附是一种非常有效的性能调控方式.CrI3单层作为一种铁磁半导体材料, 其半导体和铁磁性特征是我们重点关注的.原子的吸附是否能够对其半导体特性和铁磁特性产生影响将直接关系到该类材料的应用.通常情况下, 我们既希望保持铁磁半导体的铁磁性和半导体特性, 同时又希望调控其磁性大小.为此, 研究CrI3单层的原子吸附性质将有助于拓宽其应用领域.尽管已有文献报道了关于二维CrI3单层上Li原子吸附的结果,但未见关于其他原子吸附情况的报道.特别是, 不同类型的原子对磁性材料的影响有可能不同, 如何获得吸附原子对CrI3单层的影响规律值得我们深入研究.因此, 在本工作中, 我们研究了不同类型的原子吸附对CrI3单层电子结构和磁学性质的影响.研究的原子种类包括碱(土)金属原子Li, Mg,K; 过渡金属原子Ti, V, Mn, Fe, Co, Ni和非金属原子N, P, O, S.结果表明, 除Ti和Mn原子外的其他原子吸附不改变CrI3单层的本征铁磁属性.更重要的是, 金属原子吸附使Cr原子的局域磁矩增大, 但不超过4μB, 而非金属原子则使Cr磁矩变化呈现出多样化.我们的研究结果有助于人们对二维本征铁磁材料的磁性进行调控, 在自旋电子学中有着重要的应用前景.
2 计算方法
本文采用基于密度泛函理论的第一性原理平面波赝势方法进行研究, 所有的计算在VASP软件包[50-52]中进行.原子实和价电子之间的相互作用通过投影缀加平面波方法[53,54]描述.电子与电子间相互作用的交换关联函数采用广义梯度近似(GGA)[55]下的Perdew-Burke-Ernzerhof (PBE)[56]泛函形式.平面波基组的截断能设置为600 eV.由于广义梯度近似不能准确地描述强关联体系的电子作用, 因此本文采用GGA +U[57]的方法弛豫离子和晶胞.Cr的有效U值取为3.0 eV.该U值用于处理Cr的强局域化的d轨道电子, 与之前报道中给出的U值一致[26].布里渊区积分采用Monkhorst-Pack型网格[58],K点取值为5 × 5 × 1.为了消除周期性结构中相邻CrI3单层之间的相互作用, 我们选取20 Å的真空层.弛豫计算中总能的收敛标准为10—4eV, 每个原子的受力小于0.01 eV/Å.整个计算过程考虑了自旋极化.
3 结果与讨论
3.1 原子吸附构型
优化后的二维CrI3单层的原子结构如图1(a),(b)所示.CrI3单层由I-Cr-I三层原子构成, Cr原子层居中, 上下各有1个I原子层, 同时每个Cr原子在6个I原子所构成的八面体晶体场中.CrI3单层的原胞包含2个Cr原子和6个I原子.原胞优化后的晶格常数a为7.08 Å, 与其他报道[22]中的结果一致.
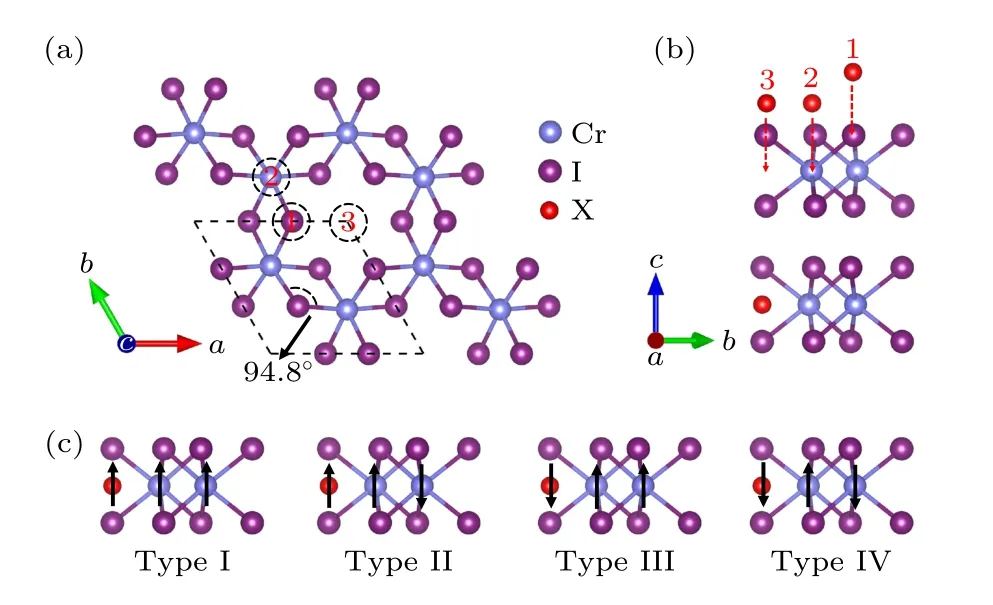
图1 (a) CrI3单层的原子结构俯视图(3个不同的吸附位用黑色的虚线圆圈表示); (b) CrI3单层的原子结构侧视图(上图中红色球为3个不同的吸附位, 下图中红色球为原子在吸附位3处吸附后的原子优化位置); (c) 四种不同的磁构型结构Fig.1.(a) Top view of atomic structure diagram of CrI3 monolayer (three different adsorption sites are represented by black dashed circle); (b) side view of atomic structure diagram of CrI3 monolayer (three different adsorption sites are shown with red balls in the top picture, and the optimized site of adsorption atom is shown with red ball in the bottom picture); (c) four different magnetic configurations.

图2 完美CrI3单层的 (a) 能态密度(红色实线表示Cr-d轨道的分波态密度, 蓝色实线表示I-p轨道的分波态密度)和 (b) 能带结构(红色实线表示上自旋电子的能带, 蓝色虚线表示下自旋电子的能带)Fig.2.(a) Energy density of states (red solid line for Cr-d projected density of states, blue solid line for I-p projected density of states) and (b) band structure (red solid line for spin up, blue dashed line for spin down) of perfect CrI3 monolayer.
完美CrI3单层是铁磁性半导体, 其能态密度和能带结构如图2(a)和图2(b)所示.在八面体晶场的作用下, Cr的5个d轨道劈裂成能级较低的t2g和能级较高的eg轨道.从图2(a)中的分波态密度可以看出, Cr自旋向上的t2g轨道为占据态, eg轨道为非占据态, 而Cr自旋向下的d轨道均为非占据态.因此, 根据Hund规则, 占据态自旋向上的t2g轨道中的3个d电子倾向于自旋平行排列,每个Cr原子具有3μB的磁矩.相邻的2个Cr原子之间通过I离子形成Cr-I-Cr超交换作用, 根据Goodenough-Kanamori-Anderson (GKA)超交换定则[59,60], 在这个体系中Cr-I-Cr的键角为94.8°,接近于90°, 因此Cr-I-Cr超交换作用为铁磁交换作用, 从而导致了CrI3体系的基态结构是铁磁性的.
为了研究吸附原子对CrI3单层性质的影响,首先要分析原子的最稳定吸附位.根据对称性要求, 我们在原胞中选取了如图1(a), (b)所示的三种吸附位进行测试.吸附位1对应于I原子的上方, 吸附位2对应于Cr原子的上方, 而吸附位3位于6个I原子形成的环中心位置.各种原子在不同吸附位的CrI3单层优化后的体系总能列于表1中, 测试结果表明, 对于每一种原子, 在位置3处吸附所形成的体系总能最低, 优化后的结构如图1(b)所示, 因此我们把位置3作为最稳定吸附位.值得注意的是, 对于金属原子吸附, 原子经过优化后嵌入到层内, 与Cr原子处于同一高度, 并且吸附原子位于I原子所形成圆环的中心位置.表1中Ti原子在吸附位1优化后与在吸附位3优化后的结构相同, 因此我们未给出在吸附位1处的体系的总能.对于非金属原子而言, P原子的情况与金属原子相似, S和N原子则会向Cr原子靠近,偏离I形成的环中心位置, 而O则会向I层靠近.由于吸附原子的引入, CrI3单层的物理性质有可能受到影响, 而产生的影响也会随着吸附原子种类的不同而不同.因此, 我们将从电子结构和磁学性质两方面对不同原子吸附后的CrI3单层进行详细研究.
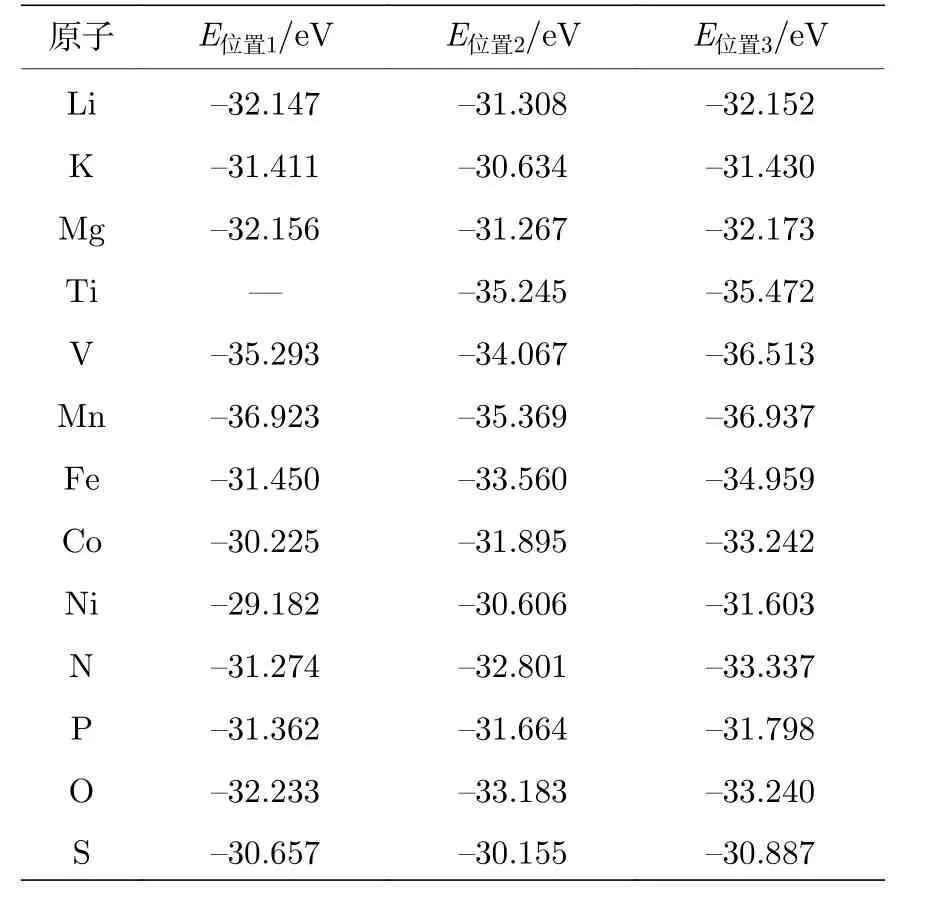
表1 各种原子位于不同吸附位的CrI3单层优化后的体系能量(E )Table 1.Energies (E ) of the optimized CrI3 monolayer with various atoms adsorbed at different sites.
3.2 碱(土)金属原子吸附的CrI3单层电子结构和磁性质
基于原子吸附的CrI3单层的最优结构, 我们需要进一步确定其磁构型.依据吸附原子和原胞中2个Cr原子的磁矩方向, 我们考虑了4种磁构型, 如图1(c)所示.其中Type Ⅰ和Type Ⅲ对应于铁磁性的CrI3单层, 而Type Ⅱ和Type Ⅳ对应于反铁磁性的CrI3单层.由于碱(土)金属原子(Li, K, Mg)不易被极化, 产生的原子磁矩可以忽略, 因此我们只考虑了CrI3单层为铁磁性和反铁磁性的两种磁构型.表2列出了计算的能量和Cr原子的磁矩.为了便于比较, 我们将基态磁构型的总能作为参考, 设定为0 eV, 而表中的数值是其他磁构型与基态磁构型的总能差值.从表2中可以明显看出, 无论是何种碱(土)金属原子吸附后, 反铁磁性的CrI3单层能量均高于铁磁构型, 意味着碱(土)金属原子吸附并未改变CrI3单层的本征铁磁性.尽管如此, Cr原子的局域磁矩却增大了.具体来说, Li和K原子吸附后, 原胞中的2个Cr原子其中的1个增大了1μB, 另1个维持不变; 而Mg原子吸附后2个Cr原子的磁矩分别增大了1μB,如表2所列.原子局域磁矩的增大意味着体系的磁性增强了.
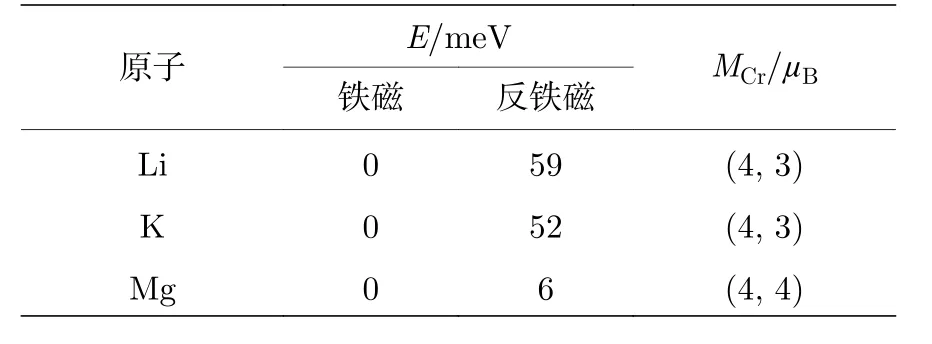
表2 碱(土)金属原子吸附后的CrI3单层的能量(E )和Cr原子的局域磁矩(MCr)Table 2.Energy (E ) and local magnetic moments (MCr)of CrI3 monolayer adsorbed by alkali (alkaline earth) metal atoms.
CrI3作为铁磁半导体材料, 不仅磁性质很重要, 半导体的特性依然是关注的焦点.为此, 我们研究了碱(土)金属原子吸附对CrI3单层电子结构性质的影响, 计算的能带结构如图3所示.从图中可以看出, 三种原子吸附后CrI3单层依然显示出半导体特性, 且带隙源自同一自旋方向.特别值得注意的是, 我们的结果与之前Guo等[49]的报道有所不同.他们发现在CrI3单层上吸附单个Li原子使体系呈现半金属化.我们仔细分析了其中的原因, 认为结果产生差异的原因主要在于他们的计算采用了GGA方法, 而对于过渡金属Cr未采用GGA +U的方法来处理局域的d轨道电子.
为了进一步分析Cr原子局域磁矩增大的原因, 我们计算了碱(土)金属原子吸附后的CrI3单层中Cr的分波态密度, 如图4所示.从图4中可以发现, 碱(土)金属原子吸附之后, Cr的态密度与吸附之前完美CrI3的情况非常相似, 不同之处在于费米能级附近的电子态发生了变化.具体来说, Li原子和K原子吸附后, CrI3单层中Cr原子在费米能级上方有2个自旋向上的非占据态, 而在费米能级下方出现1个新的自旋向上的占据态.Mg原子吸附后, Cr原子在费米能级上方有1个自旋向上的非占据态, 同时在费米能级下方也出现1个新的自旋向下的占据态.根据前面图2(a)的结果, 完美的CrI3单层中Cr原子自旋向上的eg态近似于简并, 而原胞中的两个Cr原子的态密度重合.当碱(土)金属原子吸附后, 金属原子作为电子给体, 其上的电子转移到CrI3单层上, 而Cr原子作为电子受体, 原来简并的自旋向上的eg态发生劈裂, 接受电子之后, 其中1个态成为占据态.由于碱金属原子Li和K只能提供1个电子, 从而使得其中的1个Cr原子的eg态部分成为占据态, 在这种情况下, 费米能级上方的占据态由1个Cr原子的全部eg态和另1个Cr原子的部分eg态组成.因为自旋向下的态密度在吸附前后未发生改变, 所以自旋向上的占据态增加直接导致了原子局域磁矩增加了1μB.相对于Li和K原子, Mg原子能够提供2个电子, 可以使得2个Cr原子劈裂后的eg态都成为部分占据态.在这种情况下, 2个Cr原子完全等价, 能态处于简并的状态, 故分波态密度中费米能级上方只显示1个非占据态.同样, 因为仅有自旋向上的占据态增加, 所以原子局域磁矩依然增加, 每个Cr原子分别增加1μB, 则原胞总磁矩增加了2μB.通过上面的分析可以发现, 吸附的碱(土)金属原子价电子不同, CrI3单层中磁矩增加的Cr原子数目也会不同, 因而我们可以设想通过吸附原子的种类和浓度改变CrI3单层的原子局域磁矩, 从而达到对其磁性质进行调控.
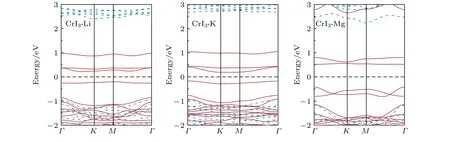
图3 碱(土)金属原子吸附的CrI3单层的能带结构(红色实线表示上自旋电子的能带, 蓝色虚线表示下自旋电子的能带)Fig.3.Band structure of the CrI3 monolayer adsorbed by alkali (alkali earth) metal atoms (red solid line for spin up, blue dashed line for spin down).

图4 碱(土)金属原子吸附的CrI3单层中Cr原子的分波态密度Fig.4.Projected density of states (PDOS) of Cr atoms in CrI3 monolayer adsorbed by alkali (alkali earth) metal atoms.
3.3 过渡金属原子吸附的CrI3单层的电子结构和磁性质
碱(土)金属原子吸附的结果相对简单, 而接下来我们将研究更为复杂的过渡金属原子(Ti, V,Mn, Fe, Co, Ni)吸附对CrI3单层性质的影响.考虑到过渡金属原子局域d轨道的强关联效应, 我们同样采用了GGA +U的方法处理过渡金属原子的3d电子, 所选用的有效U值均为3.0 eV.为了排除U值选取对结论造成的影响, 我们分别比较了U= 3.0, 4.0, 5.0, 6.0 eV时CrI3单层在吸附各种过渡金属原子后的能态密度, 发现不同U值时的计算结果极为相似, 因此我们在后续的计算中对所有的过渡金属原子都采用3.0 eV的有效U值.按图1(c)所示的4种磁构型, 我们计算了每种过渡金属原子吸附下的CrI3单层的能量, 结果列于表3.我们的结果表明, Ti和Mn原子吸附后CrI3单层的Type Ⅱ和Type Ⅳ磁构型能量相同,均低于Type Ⅰ和Type Ⅲ磁构型.V原子吸附后的CrI3单层的基态磁构型为Type Ⅰ, 而Fe, Co,Ni原子吸附后CrI3单层的基态磁构型为Type Ⅲ.由此可以看出, Ti和Mn原子吸附使CrI3单层从本征Cr-Cr铁磁序转变为反铁磁序, V, Fe, Co,Ni原子吸附不改变原有的Cr-Cr铁磁序.通过比较优化后的晶格常数, 我们发现Ti和Mn吸附之后CrI3的晶格常数分别为7.32 Å和7.25 Å, 远远大于完美CrI3的晶格常数7.08 Å, 同时也大于V,Fe, Co, Ni原子吸附后CrI3的晶格常数(V: 7.21 Å,Fe: 7.19 Å, Co: 7.16 Å, Ni: 7.12 Å).晶格常数的增大进一步导致了Cr—I—Cr键角偏离90°, 特别是Ti和Mn原子, 因此Ti和Mn原子吸附使CrI3单层发生了从铁磁向反铁磁的转变.
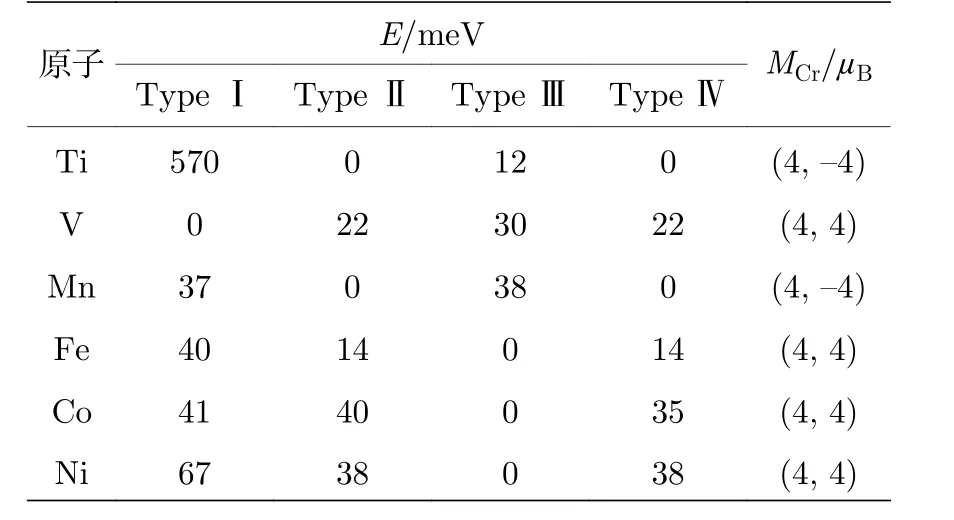
表3 过渡金属原子吸附后的CrI3单层的能量(E )和Cr原子的局域磁矩(MCr)Table 3.Energy (E ) and local magnetic moments(MCr) of CrI3 monolayer adsorbed by transition metal atoms.
同样地, 我们计算了6种过渡金属原子吸附的CrI3单层的能带结构, 如图5所示.图5中的能带结构清晰表明, 所有考虑的过渡金属原子吸附均保持了CrI3单层原有的半导体属性.CrI3单层中简并的能带在吸附金属原子后变得比较平坦.除了Ti原子吸附后的CrI3单层的带隙源于不同自旋方向, 其他5种金属原子吸附后的CrI3单层的带隙均源于同一自旋方向.与完美CrI3单层和碱(土)金属原子吸附的CrI3能带结构相比, 过渡金属原子吸附的CrI3单层的能带结构在费米能级附近变得更为复杂, 其原因是吸附原子在费米能级附近提供了更多的能态密度.

图5 过渡金属原子吸附的CrI3单层的能带结构(红色实线表示上自旋电子的能带, 蓝色虚线表示下自旋电子的能带)Fig.5.Band structure of the CrI3 monolayer adsorbed by alkali (alkali earth) metal atoms (red solid line for spin up, blue dashed line for spin down).
6种过渡金属原子吸附后的CrI3单层中Cr原子的分波态密度如图6所示.从图6可以看出, V,Fe, Co, Ni原子吸附的CrI3单层中Cr的分波态密度与碱土金属Mg原子吸附情况类似.2个等价的Cr原子劈裂后的eg态都成为部分占据态, 因此在表3中显示的V, Fe, Co, Ni原子吸附的CrI3单层中Cr原子磁矩也与Mg原子吸附体系中Cr原子磁矩相同.每个Cr原子的局域磁矩均为4μB,原胞的总磁矩增加2μB.Ti和Mn原子吸附后体系转变为反铁磁性, 如图6所示的Ti和Mn原子吸附的情况下, 除了有自旋向上的能态密度, 还有近似对称的自旋向下的能态密度, 并且Cr原子的磁矩变为4μB和—4μB, 如表3所列.值得注意的是,不同过渡金属原子的价电子数不同, 而吸附不同过渡金属原子的CrI3单层中Cr原子的局域磁矩最大值均为4μB.为了探究过渡金属原子吸附导致的Cr磁矩增加最大值为4μB的原因, 我们分析了各种过渡金属原子吸附后的磁矩值.Ti, V, Mn, Fe,Co, Ni的原子磁矩分别为2μB, 3μB, 5μB, 4μB, 3μB,2μB.结合过渡金属原子的电子排布(Ti: 3d24s2, V:3d34s2, Mn: 3d54s2, Fe: 3d64s2, Co: 3d74s2, Ni: 3d84s2)我们可以得出结论, 过渡金属原子在CrI3单层中的磁矩与孤立原子的磁矩一致, 因此过渡金属原子上只有4s电子转移到CrI3单层中, 即每个过渡金属原子只转移了2个电子, 从而导致了CrI3单层原胞中的2个Cr原子各获得1个电子, 磁矩从3μB增大为4μB.由于无进一步的电荷转移, 故Cr原子的局域磁矩最大值为4μB.结合碱(土)金属原子吸附的结果, 我们发现可以通过金属原子吸附来调控CrI3单层中Cr原子的局域磁矩.

图6 过渡金属原子吸附的CrI3单层中Cr原子的分波态密度Fig.6.Projected density of states (PDOS) of Cr atoms in CrI3 monolayer adsorbed by transition metal atoms.
3.4 非金属原子吸附的CrI3单层的电子结构和磁性质

图7 非金属原子吸附的CrI3单层的能带结构(红色实线表示上自旋电子的能带, 蓝色虚线表示下自旋电子的能带)Fig.7.Band structure of the CrI3 monolayer adsorbed by non-metal atoms (red solid line for spin up, blue dashed line for spin down).
除了金属原子之外, 我们还考虑了非金属原子(N, P, O, S)的吸附.通过测试, 我们发现非金属原子可以分为两类, 一类是易被极化的原子, 如N和S原子; 另一类是无磁矩的P和O原子.因此,对于P和O原子, 我们只考虑了Type Ⅰ和Type Ⅱ两种类型.4种非金属原子吸附的CrI3单层的磁构型的能量列于表4.从表4中发现, N, P和O 原子吸附后的基态磁构型均为Type Ⅰ.而S原子吸附的CrI3单层的基态磁构型为Type Ⅲ.4种非金属原子吸附后的CrI3单层均显示铁磁构型的能量低于反铁磁构型.

表4 非金属原子吸附后的CrI3单层的能量(E )和Cr原子的局域磁矩(MCr)Table 4.Energy (E ) and local magnetic moments (MCr)of CrI3 monolayer adsorbed by non-metal atoms.
4种非金属原子吸附的CrI3单层的能带结构如图7所示.从图7中我们可以发现非金属原子的吸附并未改变CrI3单层的半导体特性.P原子吸附虽然保持了CrI3单层铁磁半导体的特性, 但其带隙非常小.值得注意的是, N和O原子吸附时,费米能级上方的能带结构与完美CrI3单层的能带结构相似, 而P和S原子吸附时则变得较为复杂,这与非金属原子与CrI3单层耦合的程度有关.
为了分析原子吸附对Cr的局域磁矩的影响,我们计算了非金属原子吸附的CrI3单层中Cr原子的分波态密度, 如图8所示.N和O吸附后Cr的分波态密度在2个自旋方向上都与完美CrI3的相似, 因此表4中计算出的N和O原子吸附后CrI3单层中Cr原子的局域磁矩也为3μB.S原子吸附后的Cr局域磁矩为4μB, 其分波态密度与Mg原子吸附时的相似.P原子吸附后Cr原子磁矩增大至4.5μB, Cr的分波态密度清晰地显示出的费米能级附近自旋向上的占据态增加.根据前面金属原子吸附的结果, 我们可以知道原子吸附导致CrI3单层中Cr的局域磁矩增加与吸附原子和Cr原子间的电荷转移有关.如果原胞中2个Cr原子的磁矩都增加为4μB, 则表明吸附原子有2个电子转移到了Cr原子上.在Mg原子和过渡金属原子吸附时, 都只有2个s电子发生转移, 所以Cr的磁矩都只增大为4μB.P原子吸附导致的Cr原子地局域磁矩增大为4.5μB, 表明有多于2个的电子转移到了Cr上, 这与图8中的P原子吸附的CrI3单层中Cr的分波态密度是一致的.由于P上发生转移的是p电子, 其局域程度低于过渡金属中的d电子, 因而可以发生超过2个电子的电荷转移, 为此Cr的磁矩也超过了4μB.但在N, P, O, S 4种非金属原子中, P的电负性是最小的, 因而作为电子给体转移的电荷最多, 而N, O的电负性相对较大, 很难作为电子给体发生电荷转移, 所以在这种情况下Cr的磁矩维持3μB不变.

图8 非金属原子吸附的CrI3单层中Cr原子的分波态密度Fig.8.Projected density of states (PDOS) of Cr atoms in CrI3 monolayer adsorbed by non-metal atoms.
4 总 结
通过第一性原理计算, 我们将碱(土)金属原子(Li, K, Mg), 过渡金属原子(Ti, V, Mn, Fe, Co,Ni)和非金属原子(N, P, O, S)吸附在CrI3单层上, 系统地研究了原子吸附后的CrI3单层的的电子结构和磁学性质.结果显示, 金属原子倾向于吸附在6个I原子形成的环中心并且与Cr原子处在同一高度, 非金属原子吸附在I原子形成的环内,但位置依赖于原子种类.在我们所考虑的吸附原子中, 只有Ti和Mn的吸附改变了CrI3单层的铁磁性, 使其成为反铁磁半导体, 而其他原子的吸附则保持了CrI3单层的铁磁性和半导体特性.不仅如此, 我们还发现吸附不同的原子能调控Cr原子的局域磁矩.金属原子吸附使Cr原子的局域磁矩增加, 但最大不超过4μB, 而非金属原子吸附则使得结果较为复杂.O和N的吸附不改变Cr原子的局域磁矩, 而P和S原子吸附则使Cr原子局域磁矩增加, 其中 P原子吸附使Cr原子的局域磁矩增大至4.5μB.我们的研究结果为铁磁半导体CrI3单层的性能调控提供了新的思路.
猜你喜欢
数学物理学报(2022年5期)2022-10-09
华北地震科学(2022年3期)2022-07-22
银潮(2021年12期)2022-01-18
航天器工程(2019年3期)2019-07-31
黑龙江电力(2017年1期)2017-05-17
中国测试(2016年3期)2016-10-17
学生天地·小学低年级版(2016年9期)2016-05-14
上海航天(2014年1期)2014-12-31
浙江电力(2014年6期)2014-01-27
遵义师范学院学报(2010年3期)2010-09-01
