
巨噬细胞鸟苷酸结合蛋白5在新孢子虫感染中的作用
2021-06-17 12:57陈梦阁王晓岑张西臣宫鹏涛李建华
中国兽医学报 2021年5期
陈梦阁,李 新,张 旭,王晓岑,张西臣,宫鹏涛,李建华
(吉林大学 动物医学学院,吉林 长春 130062)
新孢子虫是近年来发现的一种在细胞内寄生的顶复门原虫,主要是犬新孢子虫(Neosporacaninum)。其终末宿主为犬、狼等犬科动物,中间宿主为牛、羊、鹿等多种动物,其中对牛的危害最为严重,是造成奶牛流产的主要原因之一,给养殖业造成严重的经济损失[1]。新孢子虫病呈世界性分布,包括中国在内的30多个国家和地区都有报道,我国山东、河北、内蒙古等3地奶牛的血清抗体阳性率分别为17.0%,37.0%,3.3%[2]。近年来对新孢子虫病流行病学、侵入机制等的研究很多,但依旧尚未有防治新孢子虫病的特效药及疫苗。先天免疫和获得性免疫在清除新孢子感染中发挥重要作用,对抗新孢子虫免疫机制的研究将为该病防控提供理论依据。
鸟苷酸结合蛋白(guanylate-binding protein,GBP)是一个高度保守的IFN诱导的GTPase家族,它们最初在干扰素处理的人和小鼠细胞系中被鉴定为67 kDa蛋白[3]。GBPs家族由7个人类GBPs、11个小鼠mGBPs和2个编码mGBPs的小鼠假基因组成[4],是细胞自主免疫的重要组成部分,作为对IFN信号的响应,GBPs在免疫细胞和非免疫细胞的细胞质中表达,并能迅速组装成大型防御复合体,靶向细胞内细菌、病毒和寄生虫,或释放杀伤性成分,对抗各种病原[5]。研究发现,GBPs蛋白家族在IFN-γ刺激、单核增生李斯特菌以及弓形虫感染后高表达[6],可以控制小鼠体内牛分枝杆菌[7]、嗜肺军团菌[8]和弓形虫[9]等病原感染。作为GBPs家族的成员,GBP1影响沙眼衣原体在巨噬细胞内的复制[10]、控制弓形虫的细胞内增殖[9]、并可抑制水泡性口炎病毒和心肌炎病毒的复制[11]。GBP2和GBP5抑制寨卡病毒、甲型流感和艾滋病病毒增殖[12]。GBP3抑制流感病毒的细胞内复制[13]。此外,GBP1在沙门菌感染期间促进焦亡,在弓形虫感染期间促进凋亡[14];GBP5在响应细菌感染时促进NLRP3炎症小体激活[15]。在新孢子虫感染过程中IFN-γ能够有效控制其复制和介导宿主反应[16],但是新孢子虫感染对GBPs表达的影响,以及GBPs在新孢子虫感染中的作用目前尚未见报道。
本研究首先筛选出新孢子虫感染小鼠巨噬细胞后上调表达的mGBP5基因,然后过表达mGBP5来探究其对新孢子虫生长的影响,为以GBP5为靶标开展新孢子虫病防治提供新思路。
1 材料与方法
1.1 虫体、细胞和实验动物新孢子虫速殖子Nc-1株、Vero和293T细胞均由本实验室冻存。6~8周龄野生型C57BL/6雌鼠购自辽宁长生实验动物中心。
1.2 主要试剂RPIM-1640培养基购自Life Technologies公司;胎牛血清FBS购自BI公司;Percoll购自GE Healthcare公司;20×PBS购自上海生工;巯基乙酸培养基购自BD公司;TRIzol购自Life Technologies公司;反转录试剂盒、T4连接酶均购自TaKaRa公司;FastStart Universal SYBR Master购自Roche公司;RIPA细胞裂解液购自碧云天公司;兔抗GBP5、羊抗兔IgG-HRP、羊抗兔IgG(H+L)荧光抗体均购自Proteintech公司;PCR产物回收试剂盒购自上海生工生物工程股份有限公司;质粒小提试剂盒及无内毒素质粒提取试剂盒购自天根生化生物有限公司。
1.3 新孢子虫速殖子的培养将新孢子虫速殖子接种于Vero细胞中进行培养,所用培养基为含有2% FBS和1%双抗的RPIM-1640培养基,在37℃、5% CO2细胞培养箱中进行培养,每天更换培养基,待速殖子从细胞中完全溢出后,吸出1 mL进行传代培养,其余虫体用Percoll法纯化待用。
1.4 小鼠腹腔巨噬细胞的分离及培养用3 mL巯基乙酸培养基对小鼠进行腹腔注射,3~4 d后安乐处死小鼠,将其置于75%酒精中浸泡10 min,随后在无菌条件下进行腹腔巨噬细胞的分离。用10 mL注射器将适量1×PBS注入小鼠腹腔内,随后吸出放于50 mL离心管,400 r/min离心10 min,用1 mL 培养基重悬巨噬细胞,计数后铺于细胞培养板,用添加10% FBS和1%双抗的RPIM-1640培养基于37℃、5% CO2培养箱中培养。
1.5 荧光定量PCR检测mGBPs mRNA的表达将小鼠巨噬细胞以3×106个/孔的密度铺于6孔细胞培养板培养6 h,待其贴壁,用感染复数(MOI)分别为1,3,5的新孢子虫感染细胞12 h。用感染复数为3的新孢子虫刺激细胞不同时间(6,12,24 h)收集细胞。用TRIzol法裂解细胞后,提取细胞的总RNA;将各组细胞的2 μg 总RNA利用反转录试剂盒反转录成cDNA。根据GenBank已公布序列,设计小鼠mGBP1-mGBP11和内参GAPDH荧光定量PCR的引物(表1),按照荧光定量PCR反应体系:10 μL SYBR Green Master(2×)、上下游引物(10 μmol/L)各1 μL、8 μL 5倍稀释后的cDNA,将上述几种成分避光加入到八连管中,每个样品需重复3次。反应条件为95℃,3 min→95℃,30 s;55℃,30 s;72℃,30 s(45个循环)→72℃,10 min。

表1 小鼠GBP1-GBP11 荧光定量PCR引物
1.6 Western blot检测mGBP5表达将小鼠巨噬细胞以3×106个/孔的密度铺于6孔细胞培养板,培养6 h使其贴壁,用感染复数为1,3,5的新孢子虫刺激细胞12 h,收取全细胞裂解液,使用BCA法检测蛋白含量;每孔上样30 μg,SDS-PAGE凝胶电泳进行蛋白分离,湿转法将蛋白转移至PVDF膜上,5%脱脂奶粉室温封闭2 h;4℃过夜孵育兔抗mGBP5(1∶2 000)和GAPDH(1∶1 000),TBST洗膜4次,10 min/次;结合羊抗兔IgG-HRP抗体(1∶5 000)室温孵育1 h,TBST洗膜4次,10 min/次;膜通过增强化学发光液(ECL)和Western blot检测系统进行蛋白检测,应用Image J进行灰度分析。
1.7 免疫荧光检测mGBP5定位将小鼠巨噬细胞以5×105个/孔的密度铺于24孔细胞培养板内的玻片上,待其贴壁,用新孢子虫刺激12 h,去除细胞培养上清,用1×PBS洗细胞3次;用4%的组织固定液于室温下对细胞固定20 min,洗细胞3次;随后用0.5% 的TritonX-100透化细胞20 min,洗细胞3次;之后用3%的BSA对细胞室温封闭1 h;mGBP5抗体4℃孵育过夜,用PBST洗细胞3次;再用荧光二抗对细胞进行室温避光孵育1 h,PBST洗细胞3次;用DAPI对细胞核进行染色20 min,PBST洗细胞3次;将细胞爬片晾干,用防荧光淬灭剂封片,通过激光共聚焦显微镜观察。
1.8 pcDNA3.1-mGBP5真核表达质粒的构建及转染根据mGBP5基因序列(NM_153564.2)和真核表达载体pcDNA3.1的多克隆位点设计引物,GBP5F:5′-GGGTACCGCCACCATGGCCCCAGA-GATTCACATGC-3′;GBP5R:5′-CCTCGAGGCTTATAACACAGTCATGATGATGT-3′,引物由库美生物科技有限公司合成。以小鼠腹腔巨噬细胞的cDNA为模板,PCR扩增GBP5基因。PCR产物经1%琼脂糖凝胶电泳检测后,用PCR产物回收试剂盒对所扩增的GBP5基因进行纯化。将mGBP5基因的纯化产物和pcDNA3.1质粒用KpnⅠ和XhoⅠ快切酶进行37℃酶切20 min,酶切产物经纯化回收后利用T4连接酶于4℃过夜连接;连接产物转化进入BL21感受态中,37℃摇床培养1 h后涂布于含有氨苄青霉素的LB固体培养基上,37℃倒置培养;挑取单菌落进行PCR验证,对阳性菌液扩大培养后提取质粒DNA,进行双酶切鉴定并送库美生物科技有限公司进行测序。分别将pcDNA3.1-mGBP5真核表达载体、空载体pcDNA3.1转染293T细胞24 h,Western blot检测mGBP5表达。
1.9 荧光定量PCR检测新孢子虫的数量将质量为1,3 μg的pcDNA3.1-mGBP5真核表达载体和空载体pcDNA3.1的无内毒素质粒分别转染293T细胞24 h后,用一定量的新孢子虫(MOI=0.5)感染上述各组细胞12 h后,收取细胞并提取细胞基因组,进行DNA浓度测定。根据新孢子虫(X84238)基因序列设计荧光定量PCR引物,上游引物:5′-ACTGGAGGCACGCTGAACAC-3′;下游引物:5′-AACAATGCTTCGCAAGAGGAA-3′,引物由库美生物科技有限公司合成。将1×108新孢子虫速殖子的基因组 DNA稀释成1×107,1×106,1×105,1×104,1×103新孢子虫所对应的质量200 ng,用于制作标准曲线。以试验组细胞的 200 ng 基因组 DNA 为模板,按照荧光定量PCR反应体系:10 μL SYBR Green Master(2×)、上下游引物(10 μmol/L)各1 μL、200 ng DNA、补足ddH2O(总体积20 μL)。将上述几种成分避光加入到八连管中,每个样品需重复3~4次。反应条件为95℃,5 min→ 95℃ 30 s,60℃ 30 s(30个循环)。对数据进行分析处理,根据标准品Ct值及对应的虫体数量制作标准曲线,得出试验组对应的虫体数量。

2 结果
2.1 新孢子虫感染促进小鼠巨噬细胞中GBP1-GBP11 mRNA的表达新孢子虫感染小鼠巨噬细胞后,用荧光定量PCR检测mGBPs mRNA表达。结果表明,与未感染组相比,感染复数(MOI)为1,3,5时小鼠巨噬细胞中mGBP1-mGBP11 mRNA表达均升高(图1A)。感染复数为3的虫体感染6,12,24 h 后mGBP1-mGBP11 mRNA的表达量均明显升高(图1B),其中mGBP5 mRNA表达极显著性升高(P<0.001)。

A.不同数量新孢子虫感染巨噬细胞中mGBPs mRNA表达;B.新孢子虫感染不同时间巨噬细胞中mGBPs mRNA表达。* P<0.05;** P<0.01;*** P<0.001。下同
2.2 新孢子虫感染引起小鼠巨噬细胞中GBP5蛋白表达量升高用Western blot 检测新孢子虫感染引起小鼠巨噬细胞中GBP5的蛋白表达水平,并做灰度分析。结果表明,与未感染组相比,新孢子虫感染组的mGBP5蛋白的表达量极显著性升高,且与虫体感染数量呈正比(P<0.001)(图2)。

A.GBP5蛋白的过表达;B.灰度分析
2.3 新孢子虫感染小鼠巨噬细胞中GBP5聚集在纳虫泡膜间接免疫荧光染色后通过激光共聚焦显微镜观察mGBP5在新孢子虫感染小鼠巨噬细胞中的定位。结果表明,新孢子虫感染细胞中mGBP5蛋白聚集在新孢子虫的纳虫泡膜上,而未感染组mGBP5蛋白散布在细胞质中(图3)。
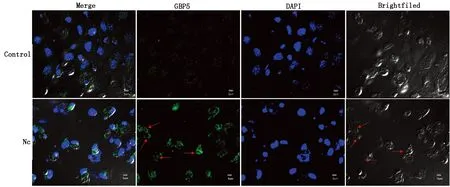
图3 间接免疫荧光观察GBP5定位(放大倍数为60倍,红色箭头指向纳虫泡)
2.4 pcDNA3.1-mGBP5真核表达载体的构建及验证琼脂糖凝胶电泳结果显示mGBP5基因扩增产物大小约为1 773 bp,与预期目的基因片段大小相一致(图4A)。重组质粒的双酶切鉴定结果表明,酶切产物大小与基因预期大小相符合(图4B)。进一步测序结果表明成功构建了pcDNA3.1-mGBP5真核表达载体。通过Western blot 检测确定pcDNA3.1-mGBP5真核表达质粒在293T细胞内成功表达,mGBP5蛋白表达量随质粒转染质量的增加而升高(图4C)。

A.PCR产物电泳图(M1.DL2000 DNA Marker;1.mGBP5 PCR产物);B.表达质粒双酶切电泳图(M2.DL10000 DNA Marker;1.pcDNA3.1-mGBP5双酶切产物);C.mGBP5蛋白过表达水平
2.5 mGBP5抑制新孢子虫的增殖新孢子虫感染分别转染pcDNA3.1-mGBP5和pcDNA3.1的293T细胞,荧光定量检测新孢子虫的数量。结果表明,与空载体组相比,mGBP5过表达组的新孢子虫数量明显减少,空载体组的虫体数量是转染GBP5(1 μg)组虫体数量的1.9倍、转染GBP5(3 μg)组虫体数量的2.8倍,且转染GBP5(3 μg)组虫体数量小于转染GBP5(1 μg)组(P<0.05,P<0.01)(图5)。以上结果显示,mGBP5蛋白起到抑制新孢子虫增殖的作用。

图5 荧光定量PCR检测新孢子虫数量
3 讨论
最近的研究表明,病毒、细菌和寄生虫等多种病原体感染都能够引起GBPs的表达上调,且GBPs与细胞膜之间的相互作用是抗病原体的机制之一。在病毒方面,已报道甲型流感病毒通过激活NF-кB途径上调GBP5的表达[17]。此外,在IFNγ刺激BMDMs中,GBP1、GBP2和相关成员IRGA6被发现定位于诺如病毒膜上[18]。在细菌方面,弗朗西斯杆菌感染小鼠骨髓源巨噬细胞上调GBP2和GBP5的表达,且共聚焦显微镜观察显示,GBP2和GBP5都定位于弗朗西斯杆菌的表面,使菌体质膜破裂,导致细菌的死亡[19]。在寄生虫方面,弓形虫弱毒株ME49和强毒株BK感染星形胶质细胞后,mGBP1、mGBP2、mGBP3、mGBP6、mGBP7和mGBP9能够聚集在弓形虫ME49株的纳虫泡膜上;而强毒株BK则能够避免mGBPs在其纳虫泡膜上的积累[6]。mGBP1在弓形虫感染的间充质基质细胞中高表达,且定位于弓形虫纳虫泡膜[20]。而mGBP2不仅被招募到弓形虫的纳虫泡膜上,而且能够进入纳虫泡腔,并直接聚集在寄生虫的质膜上,破坏了寄生虫的完整性[21]。本研究发现新孢子虫感染能够诱导小鼠巨噬细胞GBP1-GBP11共11种mGBP的mRNA表达水平升高,其中mGBP5蛋白呈高表达且聚集在新孢子虫的纳虫泡膜上。本研究还发现其他10种GBP mRNA表达也上调,它们在新孢子虫感染细胞中的定位有待下一步研究。
已报道GBPs能够抑制病毒、细菌和寄生虫等多种病原体的复制。在病毒方面,在感染脑心肌炎病毒的HeLa细胞和小鼠成纤维细胞中,mGBP2已经被证明介导抗病毒活性[22]。在细菌方面,GBP5促进NLRP3炎症小体对鼠伤寒沙门菌的抑制作用,Gbp5-/-小鼠在体外发现明显的Caspase-1和IL-1β切割缺陷,在体内发现NLRP3依赖的炎症反应和宿主防御受损[15]。在寄生虫方面,之前的研究表明mGBP1、mGBP2、mGBP3、mGBP6、mGBP7和mGBP9在IFN-γ激活的小鼠胚胎成纤维细胞中被招募到含有弓形虫的纳虫泡膜中,抑制弓形虫的增殖[6]。本研究通过分析发现小鼠与人源GBP5同源性达97%,并成功构建了过表达mGBP5的293T细胞模型,发现293T细胞中过表达mGBP5组与空载体组相比,新孢子虫数量显著减少,且与过表达mGBP5的量呈反比,表明mGBP5可抑制新孢子虫的增殖。进一步通过基因敲除等技术研究GBP5抗新孢子虫感染的作用及机制将促进我们对抗新孢子虫感染免疫应答的深入理解。
综上,本研究发现新孢子虫感染能够诱导mGBP1-mGBP11 mRNA的表达量上调,其中mGBP5蛋白表达量极显著升高。进一步以mGBP5在新孢子虫感染中的作用为主线,成功构建了pcDNA3.1-mGBP5真核表达载体及过表达mGBP5的293T细胞模型,并且发现mGBP5能够靶向新孢子虫的纳虫泡膜起到抑制新孢子虫增殖的作用。本研究为后续深入研究mGBP5的功能奠定基础,但仍需进一步研究GBPs与新孢子虫感染之间的关系,如mGBP5是否介导NLRP3炎症小体的激活来调控新孢子虫的增殖以及其他10种GBP在新孢子虫感染中的作用,这将为新孢子虫病的防治提供新思路。
猜你喜欢
现代畜牧科技(2021年3期)2021-07-21
中国预防兽医学报(2020年4期)2020-01-15
家庭百事通·健康一点通(2019年8期)2019-08-29
食品科学(2018年10期)2018-05-23
渔业致富指南(2016年12期)2016-11-11
科学大众(中学)(2015年9期)2015-10-12
西南医科大学学报(2015年1期)2015-08-22
河南畜牧兽医(2015年13期)2015-08-15
少儿科学周刊·少年版(2015年3期)2015-07-07
中国当代医药(2015年9期)2015-03-01
