
猪伪狂犬病病毒gE37~243蛋白的原核表达及gE蛋白抗体间接ELISA的建立
2021-06-17 12:32凌蒙蒙孙艺学邓效禹张鹏举丛彦龙
中国兽医学报 2021年5期
凌蒙蒙,孙艺学,邓效禹,王 杨,张鹏举,丛彦龙*
(1.吉林大学 动物医学学院,吉林 长春 130062;2.长春大学 政策法规办公室,吉林 长春 130022;3.中国农业科学院 特产研究所,吉林 长春 130112;4.吉林省农业科学院 动物生物技术研究所,吉林 长春 130033)
伪狂犬病(pseudorabies,PR)是一种急性、高度接触性传染病,其病原为伪狂犬病病毒(pseudorabies virus,PRV)[1]。1813年,美国首次在牛身上发现了一种以强烈瘙痒为特征的疾病,1849年瑞士也出现了类似的疾病,由于牛和犬感染该病后症状相似,被误认为是狂犬病[2]。直到1902年匈牙利才将该病与狂犬病区分开来,并将其命名为伪狂犬病(PR)[3]。PR自1813年发现以来,严重危害全球养猪业的健康发展。利用改良的基因缺失活疫苗免疫以及ELISA检疫等综合防控措施,美国和一些欧洲国家已经将PR消灭[4]。1947年,在我国首次发现PR[5]。由于使用了Bartha-K61疫苗,在过去30年里PR在我国的防控工作取得了巨大的成功。然而在2011年底,我国自接种了Bartha-K61疫苗的猪群中分离到了PRV[7]。此次PR疫情在各地迅速蔓延,给我国养猪业造成了沉重的经济打击。人工感染试验表明,此次新出现的病毒比以前已知的病毒毒力更强。
PRV编码至少10种糖蛋白[8],其中gE蛋白是一种囊膜糖蛋白,其参与病毒的传播并促进PRV的释放。有研究表明,缺失gE基因的PRV毒株对猪中枢神经系统中的嗅觉和三叉神经的二级和三级神经元的感染能力降低。gE蛋白也是PRV毒力的决定因子之一,但它并不是病毒体外复制所必需的蛋白。所以当gE基因缺失时,并不影响PRV的复制和抗原性,但其毒力大大降低[9]。因此目前大部分商品化疫苗均采用缺失gE基因的活疫苗。由于自然PRV毒株中都存在gE基因,因此动物感染野毒后能检测到gE蛋白的抗体。以上这些特点使gE成为PR血清学诊断的理想靶标。gE缺失疫苗免疫结合gE抗体的检测是全球公认的净化PR的根本方法。但我国PRV gE抗体检测试剂盒仍然依赖进口,还没有研发出基于本国国情的具有自主知识产权的试剂盒。基于此,本研究利用大肠杆菌表达系统表达了PRV gE的优势抗原表位,并以其为包被抗原建立了检测PRV gE抗体的间接ELISA方法,从而为鉴别PRV野毒感染与疫苗免疫提供技术支持。
1 材料与方法
1.1 毒株、载体、菌种及血清猪PRV汤阴株、pET-32a(+)载体由实验室保存;感受态细胞Rosetta(DE3)购自北京天根生化科技有限公司;PRV、PRRSV、CSFV和PCV2等阴阳性血清由河北省某养猪场提供,其中PRV、PRRSV和CSFV阴阳性血清经IDEXX ELISA试剂盒鉴定,PCV2阴阳性血清经北京百奥莱博科技有限公司ELISA试剂盒鉴定。
1.2 主要试剂2×Pfu PCR MasterMix、DNA胶回收试剂盒购自Axygen公司;DL2000 DNA Marker购自TaKaRa公司;SalⅠ、KpnⅠ购自NEB公司;T4DNA Ligase购自Thermo公司;质粒小提试剂盒购自北京天根生化科技有限公司;抗His标签鼠单克隆抗体购自北京百奥莱博科技有限公司;HPR标记羊抗猪IgG购自Solarbio公司;牛血清白蛋白(BSA)购自Seebio公司;显色液TMB购自北京梅科万德有限公司。Ni-NTA Agarose购自Qiagen公司;PRV gE糖蛋白阻断ELISA抗体检测试剂盒购自西班牙海博莱生物大药厂。
1.3 目的基因扩增及表达质粒构建根据gE基因序列,设计了1对扩增gE37~243的引物。引物序列如下:gE37~243-F:5′-GGGGTACCGAGGTTCCATCCCCATCTG-3′;gE37~243-R:5′-ACGCGTCG-ACGAGAATTTCACTGCCACC-3′。用T4连接酶将酶切后的目的片段与pET-32a(+)载体在25℃下进行连接。30 min后,将连接产物转化至DE3感受态细胞。涂板后挑取单菌落进行摇菌,经菌液PCR鉴定正确后提取质粒并测序,将测序正确的质粒命名为pET-gE37~243。
1.4 重组蛋白表达将pET-gE37~243培养菌液接种于含Cmr+和Amp+的LB液体培养基,37℃ 180 r/min振荡培养至D600=0.6~0.8,取出200 μL菌液作为对照,而在剩余菌液中加入终浓度为0.25 mmol/L 的IPTG进行诱导。 3 h左右时取出200 μL诱导菌液,13 000 r/min离心1 min,弃上清。用40 μL PBS重悬沉淀后,加入8 μL 5×loading Buffer,沸水煮5~10 min后, 12 000 r/min离心3 min,取上清进行SDS-PAGE电泳,观察蛋白表达情况。
1.5 重组蛋白纯化与Western blot鉴定根据Ni-NTA Agarose说明书对目的蛋白进行纯化,纯化后的蛋白于1×PBS中4℃透析过夜。透析后的蛋白先经SDS-PAGE电泳,然后转移至硝酸纤维素膜上进行Western blot鉴定。
1.6 间接ELISA方法的建立
1.6.1抗原包被质量浓度及最佳样品稀释倍数的确定 通过棋盘法确定抗原包被浓度以及被检血清样品的最佳稀释度,具体步骤如下:用pH 9.6的碳酸盐缓冲液将0.5 mg/L的纯化蛋白稀释至0.03 125 mg/L,100 μL/孔,4℃包被过夜。用含0.1%吐温的PBS洗涤3次后加入1% BSA,37℃封闭2 h。洗版后每孔加入PBS稀释的猪血清,稀释度依次为:1∶20,1∶40,1∶80,1∶160,1∶320,1∶640,37℃孵育1 h。洗涤后加入1∶2 000倍稀释的HPR标记羊抗猪IgG酶标抗体,100 μL/孔,37℃孵育1 h。洗板后每孔加入100 μL的TMB显色液,37℃避光显色10 min。10 min后立即加入50 μL 0.05 mol/L H2SO4终止反应,然后测定D450值。
1.6.2间接ELISA检测方法判定标准的确定 采用本研究所建立的PRV抗体检测间接ELISA方法对50份标准阳性血清样本进行检测,同时检测SPF猪血清作为对照。测定D450值,经统计学分析确定ELISA检测方法的判定标准。
1.6.3敏感性和特异性检测 采用本研究所建立的检测PRV gE抗体的间接ELISA方法,检测30份阳性猪血清样本,根据检测结果评价本研究所建立方法的敏感性。此外,对32份PRRSV阳性血清、PCV2阳性血清、CSFV阳性血清以及PRV gE缺失苗免疫的猪血清进行检测,以确定所建立的方法的特异性。
1.6.4重复性检测 取同一批次以及不同批次的酶标板各6块,检测阳性样品和阴性样本,分别计算批内和批间变异系数,以评价本研究所建立方法的重复性。
1.6.5符合率检测 采用本研究所建立的间接ELISA方法检测30份阳性猪血清样本及45份阴性样本,并与PRV gE糖蛋白阻断ELISA抗体检测试剂盒进行对比,进而评价所建立方法的检测性能。
2 结果
2.1 目的基因扩增与菌液PCR鉴定将PCR扩增的gE37~243基因产物经1%琼脂糖凝胶电泳,可见约在681 bp处出现1条目的条带(图1)。pET-gE37~243菌液PCR鉴定结果显示,扩增条带与预期大小相符(图2),表明重组质粒pET-gE37~243构建正确。

M.DL2000 Marker;1.gE37~243基因

M.DL2000 DNA Marker;1.pET-gE37~243重组质粒
2.2 gE37~243蛋白纯化及Western blot分析将诱导前后的菌液进行SDS-PAGE电泳。结果显示,在菌液上清中检测到1条约45 kDa的条带(图3),表明gE37~243蛋白在菌液上清中以分泌方式表达。对gE37~243蛋白上清进行Ni-NTA纯化,得到质量浓度为42 mg/L的较纯目的蛋白(图4)。Western blot分析结果表明gE37~243蛋白可以与抗His标签单克隆抗体结合,表明目的蛋白纯化成功。

M.蛋白质相对分子质量标准;1.pET-32a(+)对照上清;2.pET-32a(+)对照沉淀;3.pET-gE37~243诱导前;4.pET-gE37~243诱导后
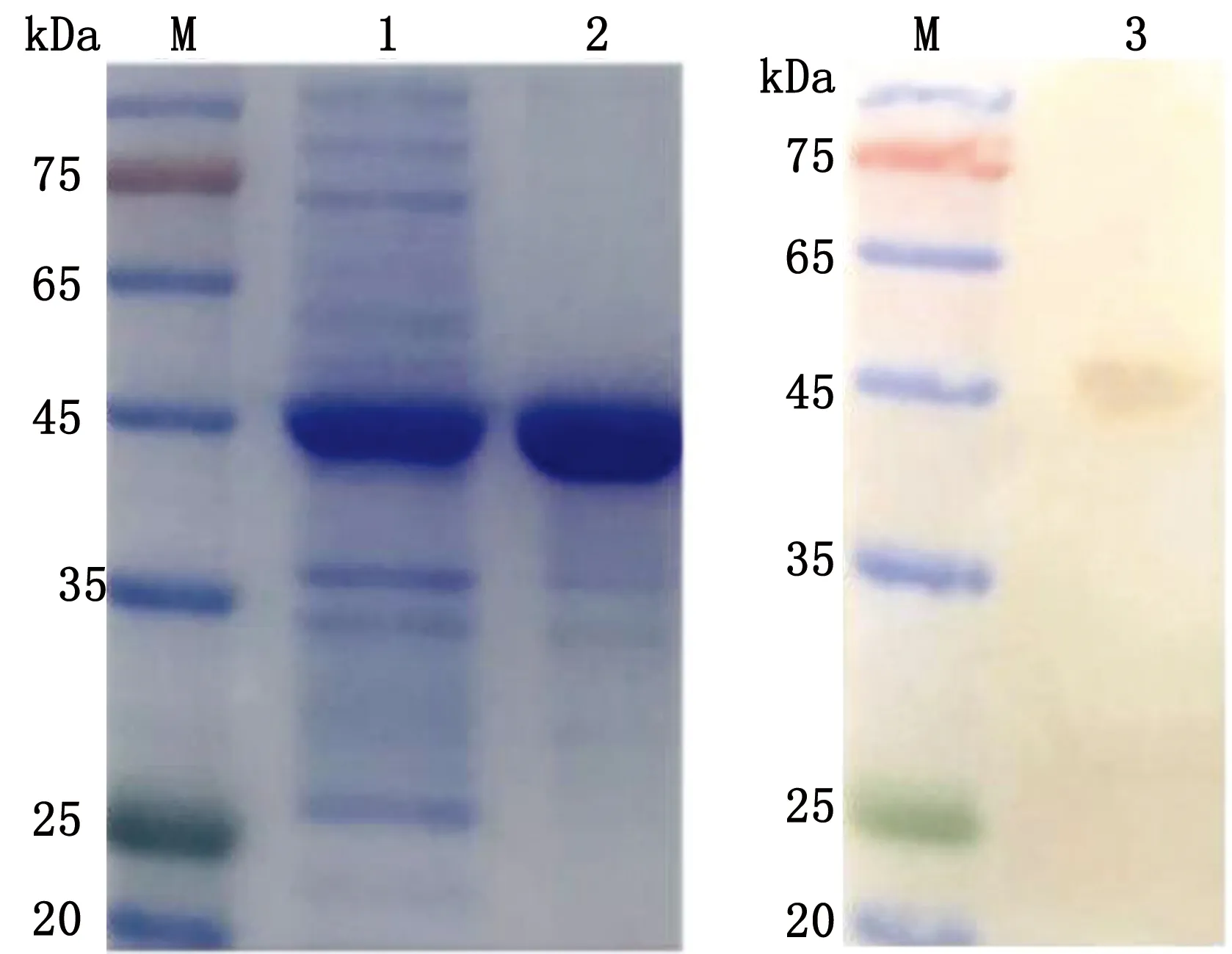
M.蛋白质相对分子质量标准;1.gE37~243蛋白纯化前;2.gE37~243蛋白纯化后;3.gE37~243蛋白与His单抗反应
2.3 间接ELISA检测方法的建立
2.3.1抗原最佳包被浓度和最佳血清样品稀释倍数的确定 由表1可知,当抗原质量浓度和血清的稀释倍数分别为0.25 mg/L和1∶80时,阳性值(P值)更接近1,阴性值(N值)小于0.3。因此抗原质量浓度是0.25 mg/L和抗体稀释倍数是1∶80是本方法的最佳工作浓度。

表1 抗原包被质量浓度和血清样品稀释倍数优化结果
2.3.2间接ELISA检测方法判定标准的确定 利用所建立的检测PRV gE抗体的间接ELISA方法对SPF猪血清和50份PRV阳性血清进行检测,发现阳性血清D值和阴性血清D值的比值(S/N)均大于2.0。据此,判定为阳性的标准为S/N>2.0。
2.3.3敏感性和特异性确定 根据敏感性计算公式Sens itivity=TP/(TP+FN)×100(TP代表真阳性,FN代表假阴性)评价本研究所建立方法的敏感性。经过计算,在对30份PRV阳性血清进行检测过程中其敏感性为93.33%。根据特异性计算公式Specificity=TN/(TN+FP)×100,对32份PRRSV阳性血清、PCV2阳性血清、CSFV阳性血清以及PRV gE缺失苗免疫的猪血清进行检测。经过计算,本方法的特异性为93.75%。
2.3.4重复性确定 利用所建立的检测PRV gE抗体的间接ELISA方法进行重复性试验,结果如表2所示。其中,批内变异系数为2.175%~9.272%,批间变异系数为2.443%~7.688%,均低于10%。由此表明该方法具有较好的重复性。

表2 重复性试验结果
2.3.5与商品化试剂盒检测结果比较 分别采用本研究所建立的检测PRV gE抗体的间接ELISA方法和PRV gE糖蛋白阻断抗体检测试剂盒同时检测30份猪血清阳性样本及45份阴性样本,并对比两者的检测结果。由符合率计算公式Coincidence=(TP+TN) / (TP+FN+TN+FP)×100计算结果可知,两种方法的符合率可达92.00%(表3)。

表3 间接ELISA和阻断ELISA对临床样本检测结果对比
3 讨论
猪PR自2011年底在我国再次暴发,已成为当前危害我国养猪业最严重的疾病之一。我国政府要求全国所有种猪场到2020年将PR达到净化标准[10]。gE缺失疫苗和gE抗体血清学检测相结合的方法是目前净化和防控PR最有效的方法。目前常用的gE抗体检测血清学方法包括乳胶凝集实验、琼脂扩散试验和ELISA等。由于ELISA方法的特异性、准确性、敏感性和检出率更高而受到检测人员的广泛关注。
PRV gE基因的开放阅读框长度约为1 731 bp,可编码577个氨基酸。研究表明,当在大肠杆菌中表达全长gE蛋白时,由于gE蛋白在内质网中的积累,抑制了gE蛋白的合成,导致很难在系统中成功表达全长gE基因[11]。因此,本研究选择在E.coliRosetta(DE3)中表达gE蛋白的主要抗原表位区,并利用获得的目的蛋白作为包被抗原建立了检测gE抗体的间接ELISA方法。结果显示,该方法的敏感性和特异性分别为93.33%和93.75%,批内和批间变异系数均低于10%,与商品化试剂盒的符合率为92.00%。由此表明,本研究所建立的检测PRV gE抗体的间接ELISA方法具有较好的敏感性、特异性和重复性,能够满足鉴别PRV野毒感染和疫苗免疫的需求,该方法为国内PR的净化提供了技术支持。
猜你喜欢
中国土壤与肥料(2021年5期)2021-12-02
疯狂英语·新悦读(2020年7期)2020-07-30
癌变·畸变·突变(2020年1期)2020-02-12
天津医科大学学报(2019年6期)2019-08-13
中国有色金属学报(2018年2期)2018-03-26
现代检验医学杂志(2016年2期)2016-11-14
焊接(2016年1期)2016-02-27
上海蔬菜(2015年2期)2015-12-26
新闻传播(2015年8期)2015-07-18
肿瘤预防与治疗(2014年2期)2014-11-24
