
多药同服对大鼠回肠P-糖蛋白的影响
2021-06-17 08:10苑键,万军
武警医学 2021年5期
苑 键,万 军
世界卫生组织将多药同服定义为“同时服用多种药物或过量服用药物”[1]。即使在许多情况下是由于伴随的疾病或者复杂的治疗情况会使用更多的药物,多药同服与多发共病的存在与药物-药物或药物-疾病的相互作用呈现高概率相关[2]。
相关研究发现,许多口服药物生物利用度低的主要原因与存在于肠黏膜的P-糖蛋白(P-gp)有关,P-gp作为一种能量依赖性药物外排泵,通过ATP供能,将细胞内的药物泵出细胞外,从而降低细胞内的药物浓度[3]。P-gp的这种作用被认为是导致口服药物生物利用度降低的主要原因。同时,在胃肠道中P-gp活性和功能的定量数据目前还不清楚[4]。
垂直扩散池(ussing chamber)是研究胃肠道屏障功能的金标准。能够模拟肠道的生理环境并对药物转运的不同阶段进行研究。根据前期调研发现,我院患者同时服用抗血小板聚集药物、抗抑郁药物、抑酸药、降糖药的占很大比重,因此选择了氢氯吡格雷、雷贝拉唑、草酸艾斯西酞普兰、甲苯磺丁脲这四种药物进行多药同服的肠道转运实验。
本研究使用大鼠离体回肠段,通过ussing chamber转运实验研究多药同服对P-gp底物罗丹明123(R123)的影响,以及非P-gp底物荧光素钠(CF)的转运行为,研究目标药物与肠道内P-gp交互作用,以了解多药同服与单药应用在肠道转运层面是否存在差异,进一步指导临床用药。
1 材料与方法
1.1 仪器与材料 Ussing chamber(北京金工鸿泰),荧光测定仪F-2000(日本东京HITACHI株式会社),罗丹明123、荧光素钠、戊巴比妥钠(Sigma公司),硫酸氢氯吡格雷标准品、雷贝拉唑钠标准品、草酸艾司西酞普兰标准品、甲苯磺丁脲标准品、维拉帕米标准品(solarbio公司),其余试剂均为分析纯。
1.2 实验方法
1.2.1 分组方法与动物模型制备 40只大鼠随机分为4组(n=10),分别为:空白对照组(生理盐水),阳性对照组(维拉帕米),单药组(氯吡格雷),多药组(氯吡格雷+雷贝拉唑+草酸艾司西酞普兰+甲苯磺丁脲),每组分别进行吸收试验(n=5)和分泌试验(n=5)。药物剂量分别为:生理盐水 2 ml/只;氯吡格雷 30 mg/kg,维拉帕米 20 mg/kg,草酸艾司西酞普兰 20 mg/kg,雷贝拉唑 20 mg/kg,甲苯磺丁脲 500 mg/kg。上述药物均用生理盐水配制,每只大鼠每日灌胃1次,溶液剂量为2 ml。灌胃7 d后每组分别调查罗丹明123和荧光素钠的肠道转运。
1.2.2 Ussing chamber实验 Wistar雌性大鼠(北京维通利华),8周龄,(230±20) g。实验大鼠给予样品1周后, 禁食16~18 h, 3%戊巴比妥钠(30 mg/kg)腹腔注射麻醉, 延腹中线将腹部切开2.5 cm左右, 胃下5 cm处为空肠(空肠约10 cm), 在空肠前端插入硅胶管(内径2 mm)结扎, 空肠末端剪开一小口后, 轻缓地用生理盐水冲洗肠段2次、通入空气4次, 分离回肠置Krebs-Ringer缓冲液中, 冰浴培养5 min,剪取3~4 cm长的肠段, 迅速分离除去筋膜层, 将肠黏膜固定于扩散池上。吸收方向时(M-S),在黏膜侧加入5 ml R123溶液或CF溶液, 浆膜侧加入5 ml Krebs-Ringer缓冲液;分泌方向实验时(S-M),浆膜侧加入5 ml的R123溶液或CF溶液,黏膜侧加入5 ml Krebs-Ringer缓冲液。在混合气体(95% O2/5% CO2)的作用以及37.5 ℃水浴保温下, 分别于15、30、45、60、75、90和120 min在接收侧取样0.5 ml, 同时补充0.5 ml 37.5 ℃ Krebs-Ringer缓冲液。各份取样后的样品于冰箱中保存直到进行荧光分析。
1.2.3 罗丹明123和荧光素钠的方法学考察 罗丹明123的激发波长为485 nm,发射波长为535 nm,可在此波长下测定其荧光强度,空白无干扰。罗丹明123在10~200 μg/L,荧光强度对浓度进行回归,Y=0.2238X+2.5657,线性关系良好(相关系数R2=0.9978),回收率和日内精密度分别为99.8%和0.8%。
荧光素钠的激发波长为480 nm,发射波长为520 nm,可在此波长下测定其荧光强度,空白无干扰。荧光素钠在200~2000 μg/L,荧光强度对浓度进行回归,Y=0.6373X+0.0699,线性关系良好(相关系数R2=0.9987),回收率和日内精密度分别为99%和2.6%。
1.2.4 累计渗透量及表观渗透系数的测定 将荧光分析法测定的每个样品的数据处理, 得到给定时间浆膜侧(吸收方向)药物质量浓度ρtn1和黏膜侧(分泌方向)药物质量浓度ρtn2 , 用公式Qtn =Σ0.5ρt(n-1)+5ρtn计算累计透过量(Qtn), 其中0.5和5表示取样体积和加入药液量,分别为0.5和5 ml。根据给药剂量D,计算累计透过百分率(Qtn/D×100%)。另外,表观渗透系数(Papp)是表示药物经黏膜透过能力的重要指标, 其计算公式为: Papp=(dQ/dt)×(1/Aρ0 ), 单位cm/s, 其中dQ/dt表示稳态时时间-累计透过量线性回归所得的斜率,A为有效渗透面积(1.78 cm2),ρ0为加入药液侧初始药物质量浓度。在本实验中, 泵出比(efflux ratios, ER)用于评价对P-gp的抑制作用效果, 其计算公式为ER=PappSM /PappMS, 其中PappSM是分泌方向的平均渗透系数, PappMS是吸收方向的平均渗透系数,其中SM为分泌方向(浆膜侧到黏膜侧转运);MS为吸收方向(黏膜侧到浆膜侧转运)。
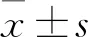
2 结 果
2.1 不同实验组对R123经回肠黏膜转运的Papp的影响 (1)吸收方向:R123经大鼠回肠黏膜吸收方向的经时曲线,阳性对照组与空白对照组相比,透过率有显著增高趋势,差异有统计学意义(P<0.05)。单药组、多药组与空白对照组相比无统计学差异。虽然多药组对R123在吸收方向较单药组有增加趋势,但是两者相比无统计学差异(图1)。阳性对照组Papp与空白对照组相比有统计学差异(P<0.05),单药组Papp与多药组相比有增高趋势,但两者相比无统计学差异(表1)。(2)分泌方向:R123经大鼠回肠黏膜分泌方向的经时曲线,阳性对照组与空白对照组相比,透过率有统计学差异(P<0.05),多药组和单药组相比,透过率有显著增高,差异有统计学意义(P<0.05)。单药组、多药组与空白对照组透过率相比无显著性差异(图1)。阳性对照组Papp与空白对照组相比有显著性差异(P<0.05),多药组Papp较单药组明显增高,差异有统计学意义(P<0.05,表1)。(3)ER:空白对照组与阳性对照组相比有显著性差异(P<0.05),单药组与阳性对照组相比有显著差异(P<0.05),多药组与单药组相比有统计学差异(P<0.05),多药组与空白对照组相比有显著性差异(P<0.05,表1)。

表1 不同实验组对罗丹明123对大鼠回肠黏膜Papp的影响
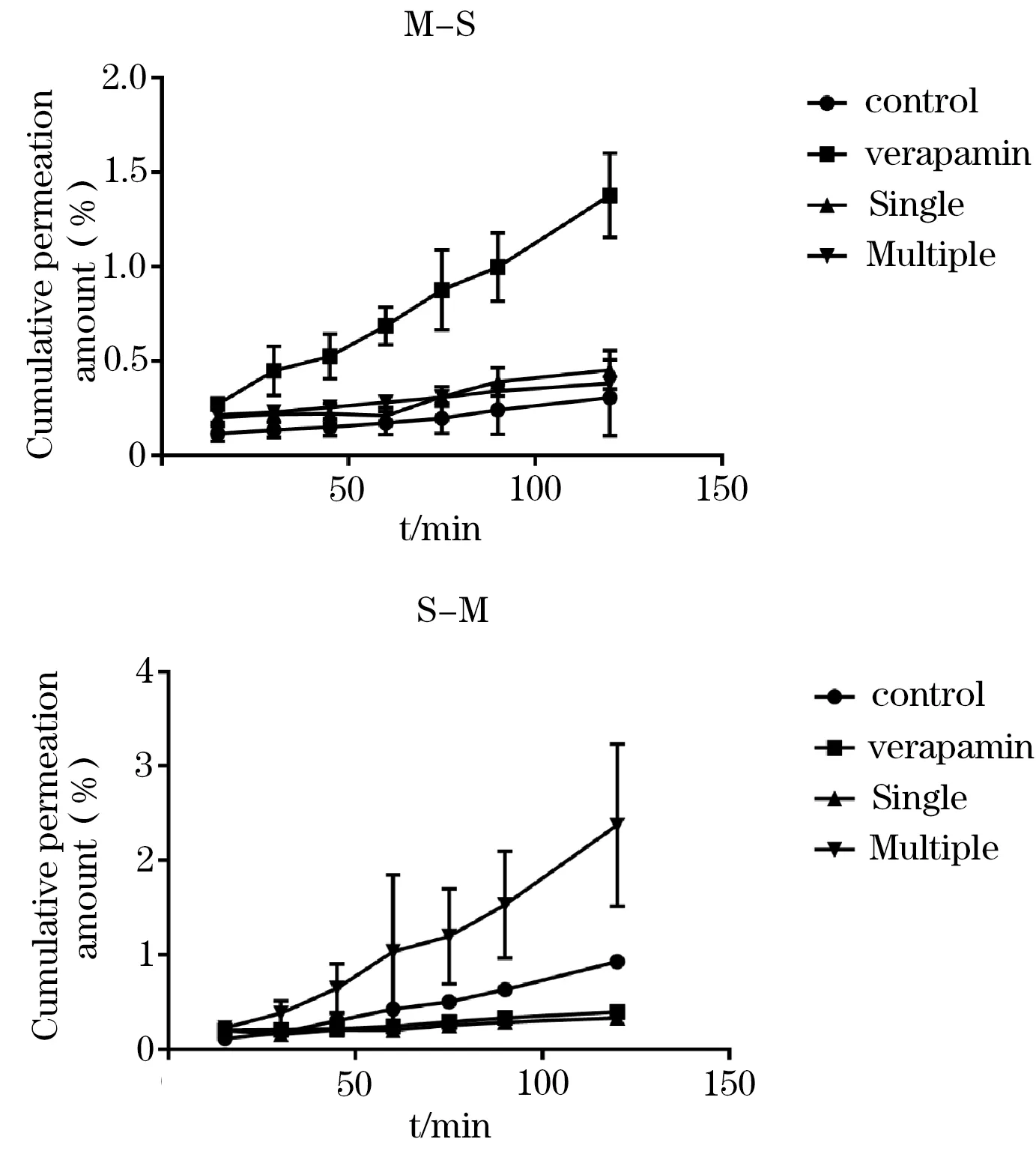
图1 各实验组对罗丹明123经大鼠回肠黏膜吸收方向透过和分泌方向透过的经时效益
2.2 不同实验组对荧光素钠经回肠黏膜转运的Papp的影响 (1)吸收方向:CF经大鼠回肠黏膜吸收方向的经时曲线,单药组、多药组分别与空白对照组相比,透过率有显著降低趋势,差异有统计学意义(P<0.05)。虽然多药组在吸收方向较单药组相比有增高趋势,但两者相比无统计学差异(图2);多药组Papp和单药组Papp相比,差异无统计学意义(表2)。(2)分泌方向:空白对照组与其余各组间相比有统计学差异(图2),各组ER相比无统计学差异(表2)。
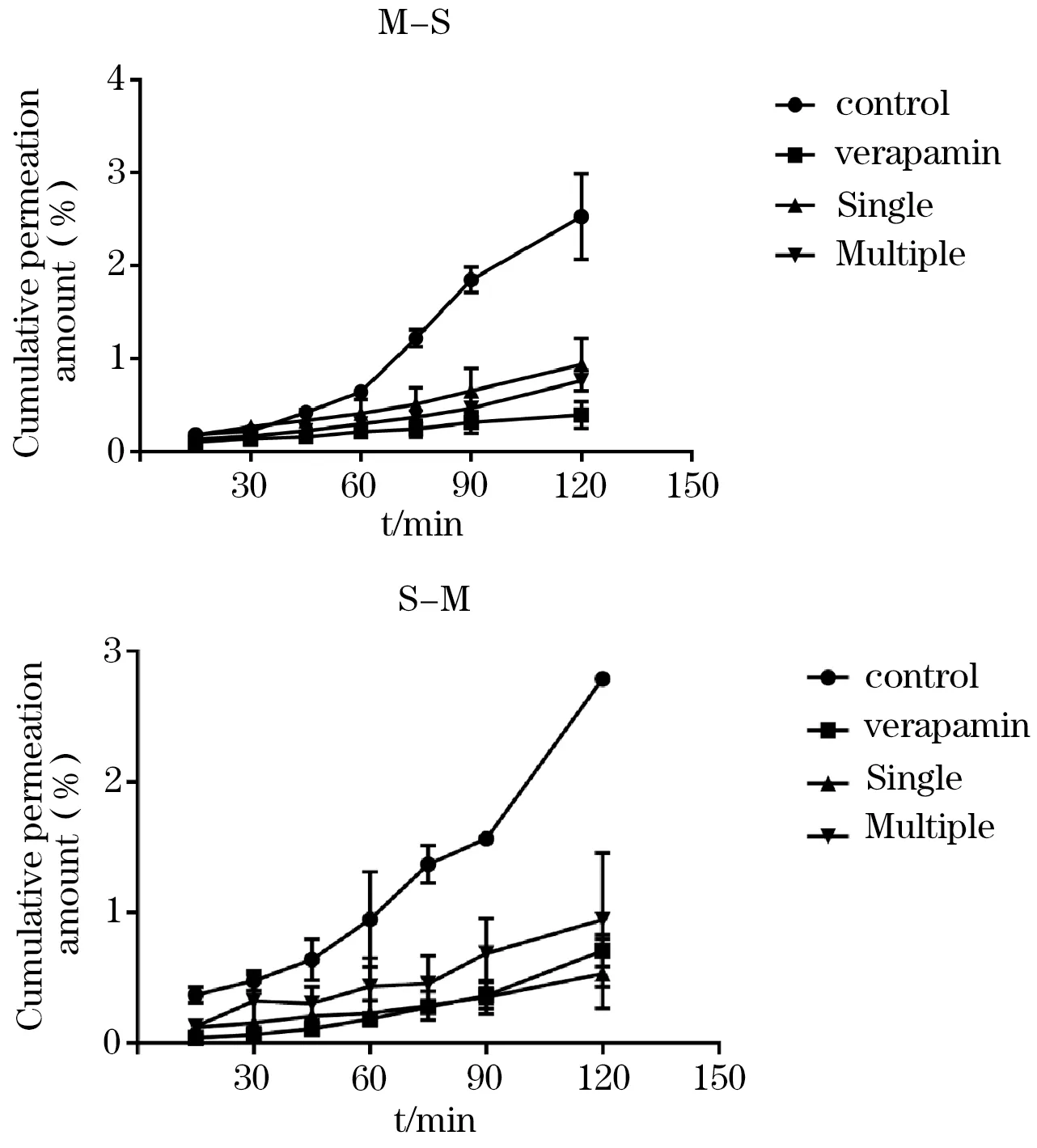
图2 各实验组对CF经大鼠空肠黏膜吸收方向透过和分泌方向透过的经时效益
表2 不同实验组CF对大鼠回肠黏膜Papp的影响
3 讨 论
影响药物吸收的主要屏障可概括为化学屏障、物理屏障和生化屏障三大类[5]。肠上皮细胞的代谢酶和药物外排泵构成是生化屏障的重要部分。代谢酶主要通过体循环前代谢将已进入肠上皮细胞的相应药物代谢,丧失其活性。药物外排泵系统主要通过P-gp、MRP、MTX等外排系统[6,7],将肠道细胞内已吸收的药物“泵”回肠腔,从而降低肠道细胞内药物浓度,导致口服药物生物利用度减弱[8]。
本研究分别给大鼠灌胃药物1周后,观察4个实验组在120 min内R123经大鼠回肠的经时透过量。研究中发现,空白对照组分泌方向透过的经时曲线与吸收方向相比有统计学差异,验证了R123是P-gp的底物,而维拉帕米(阳性对照组)逆转了这种作用,同时验证了维拉帕米是P-gp的抑制药。单药组R123的转运方向由分泌为主变为吸收为主,说明氯吡格雷可以抑制P-gp的功能(空白对照组ER=6.18±3.57vs.单药组ER=0.65±0.31,P<0.05)。多药组吸收方向累计透过率与单药组相比无统计学差异,而分泌方向的Papp有统计学差异(单药组Papp=0.86±0.18vs.多药组Papp=11.42±4.38),同时两者ER相比也有统计学意义(单药组ER=0.65±0.31vs.多药组ER=12.27±4.74)。说明药物在空肠黏膜的转运过程中,氢氯吡格雷可以抑制P-gp的功能,减少药物的分泌,而多药组会减轻这种抑制作用,使药物外排量增加,减少了药物的吸收量。同时发现多药组ER与空白对照组相比有统计学差异(多药组ER=12.27±4.74vs.空白对照组 ER=6.18±3.57),可能由于药物种类的增加增强了P-gp的功能,也可能由于P-gp的外排作用使多药在肠道中停留时间增加,产生了毒性物质所导致。
除了空白对照组使CF在吸收方向和分泌方向透过增加,其余药物均使CF的透过显著降低,说明实验药物均为载体吸收而非细胞旁路途径吸收。有研究表明,通过可逆性调节紧密连接孔径促进吸收的药物则毒性较小,这类物质曾经作为药物辅料添加在口服制剂中,然而肠上皮细胞膜暂时开放所引起的有害物质的吸收,仍是一个无法回避的安全隐患问题[9]。而本实验也验证了这一点,不论从吸收方向还是分泌方向,维拉帕米组、单药组和多药组的累计透过率和Papp均低于空白对照组。
综上所述,本实验发现,氯吡格雷可以抑制回肠P-gp的功能,但是多药同时服用会明显减弱氯吡格雷的这种抑制作用。因此,在共病治疗或者综合性治疗时,尽量避免多种药物同时服用,防止出现口服药物生物利用度降低,减弱药效,从而导致治疗失败。
猜你喜欢
中日友好医院学报(2022年4期)2022-10-15
中国药学药品知识仓库(2022年9期)2022-05-23
肝博士(2020年5期)2021-01-18
中国食用菌(2020年9期)2020-11-11
看世界·学术下半月(2020年7期)2020-09-10
医学食疗与健康(2019年6期)2019-09-10
广东农业科学(2017年10期)2018-01-25
中国医学创新(2017年22期)2017-11-15
中国继续医学教育(2015年1期)2016-01-06
西南军医(2015年1期)2015-01-22
