
3日龄男婴呼吸困难合并内脏反位
2021-06-16 02:08林慧佳罗芳马晓路
中国当代儿科杂志 2021年6期
林慧佳 罗芳 马晓路
(1.浙江大学医学院附属儿童医院新生儿重症监护室/国家儿童健康与疾病临床医学研究中心,浙江杭州 310052;2.浙江大学医学院附属第一医院儿科,浙江杭州 310003)
1 病例介绍
(1)病史:患儿男,生后3 d,因气促2 d、发绀1 d入院。2 d前(生后1 d)在当地医院母婴同室发现呼吸稍增快,1 d前气促明显,无发热,无少吃、少动,无咳嗽、抽搐等,转入当地新生儿科治疗气促无改善,伴口周发绀,为进一步治疗,转入我院。患儿系第2胎第1产,胎龄39+5周,剖宫产出生,出生体重3 340 g,Apgar评分1 min 10分、5 min 10分,羊水清,脐带、胎盘无异常。父亲33岁,母亲29岁,身体健康。母亲孕期查B族链球菌阴性;母亲既往胚胎停育1次,原因不详。否认近亲结婚,家族中无遗传性疾病及其他重大疾病史。
(2) 入 院 体 检 :T 36.7℃ ,P 150次/min,R 78次/min,BP 71/37 mm Hg, 平 均 动 脉 压46 mm Hg,鼻导管吸氧下SpO296%,反应可,哭声响亮,轻度三凹征,双肺呼吸音粗,可闻及湿啰音,心尖搏动最强点位于右侧锁骨中线第5肋间,心脏听诊右侧胸腔明显,心律齐,未闻及杂音,腹软,肝肋下1 cm,脾肋下未及,肠鸣音3~5次/min,四肢肌张力正常,生理反射可引出,毛细血管再充盈时间2 s。
(3)辅助检查:血常规示WBC 10.7×109/L(参考值:15.00×109/L~20.00×109/L),余正常。C反应蛋白(C-reactive protein,CRP):23.36 mg/L(参考值:0~8 mg/L)。血气分析示pH 7.229,PCO262.6 mm Hg,PO244.2 mm Hg。血培养、痰培养阴性。痰解脲脲原体、沙眼衣原体DNA阴性。胸腹X线片(入院第1天)示双肺纹理增多,心尖偏右,胃泡影似位于右上腹,左上腹偏密实,肝影可能(图1)。心脏超声(入院第3天)示镜像右位心,房间隔缺损。头颅B超未见异常。腹部超声示内脏反位,肝胆脾胰未见明显异常;双肾、输尿管、双侧肾上腺未见明显异常。胸部心脏大血管CT血管成像及气道重建(入院第5天)示镜像右位心伴内脏反位,房间隔缺损,右位主动脉弓(图2);双肺透亮度不对称,双肺纹理增多,模糊,肺野内散在模糊片状密度增高影,气管及支气管通畅。行床边支气管镜(入院第6天,患儿生后9 d)检查示经鼻进镜顺利,鼻腔可见大量脓性分泌物,喉、声带无异常,左右主支气管开口及各分支开口通畅,黏膜略充血水肿,各管腔内可见较多脓性分泌物,予0.9%氯化钠溶液灌洗,洗出黏液栓样液体。入院第14天自动听性脑干反应示双耳通过测试。
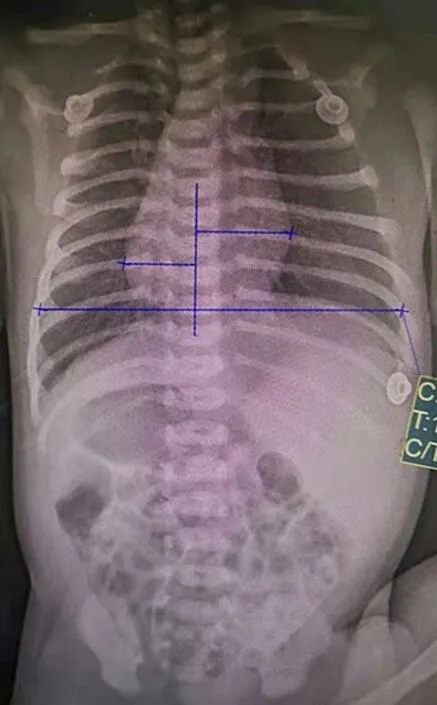
图1 患儿入院第1天胸腹X线片结果 双肺纹理增多,心尖偏右(蓝色箭头所示),胃泡影位于右上腹(红色箭头所示),左上腹偏密实,考虑肝影(黑色箭头所示)。
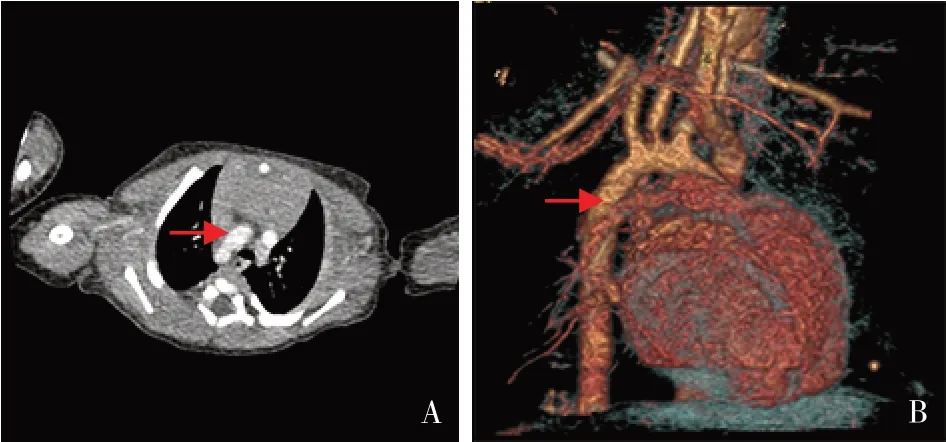
图2 患儿入院第5天胸部心脏大血管CT血管造影及气道重建结果 图A、B示右位心、右位主动脉弓(箭头所指为右位主动脉弓)。
2 诊断思维
患儿病例特点:(1)足月适于胎龄儿,急性起病;(2)生后1 d即开始出现呼吸困难、发绀症状;(3)CRP轻度升高,胸片及胸部CT提示肺野片状增高影,超声提示镜像右位心、内脏反位,支气管镜提示鼻腔大量脓性分泌物及气管腔内较多脓性分泌物。家族史无特殊。患儿为新生儿期出现的呼吸困难,结合病例特点,主要从3个方面考虑:(1)中枢神经系统疾病:该患儿出生时无窒息史,无神经系统症状,头颅B超未见异常,无缺氧、颅内出血等神经系统疾病,故不支持神经系统疾病影响呼吸中枢功能,引起呼吸症状。(2)循环系统疾病:新生儿严重的先天性或后天性心脏病、持续肺动脉高压等常伴有心力衰竭,引起肺淤血、肺顺应性下降,出现换气功能障碍,从而导致呼吸困难。该患儿虽然心脏超声提示镜像右位心,但无并发先天性心脏病、心功能异常、肺动脉高压等。另外,新生儿红细胞增多症或贫血可因缺氧引起呼吸困难,但该患儿血红蛋白正常,不支持红细胞增多症或贫血。(3)呼吸系统疾病:新生儿期出现呼吸困难最常见为新生儿呼吸窘迫综合征、新生儿湿肺、吸入性肺炎、感染性肺炎。该患儿发生呼吸困难并非生后6~12 h,胸片无细网状颗粒影、肺透亮度减低、支气管充气征等,故不支持新生儿呼吸窘迫综合征;患儿呼吸困难并非一过性、自限性,胸片也无叶间积液、云雾状阴影等,故不支持湿肺;患儿出生时无吸入病史,羊水清且无粪染,故不支持吸入性肺炎;患儿辅助检查提示CRP升高,需排除新生儿感染性肺炎,但病原体检查均阴性,治疗后虽CRP正常但气促症状无好转。另外,胸片上无气胸、纵隔气肿、心包积气、肺囊肿,无膈面抬升,胸腔未见肠管影,肺容积未见异常等,故不支持肺气漏、先天性肺囊肿、先天性肺发育不全、膈疝、膈膨升等先天性肺部疾病。胸部CT气道重建和支气管镜检查均提示支气管通畅,故不支持气管狭窄或气管畸形等。因此考虑是否存在其他特殊的呼吸系统疾病。因患儿影像学检查证实有内脏反位,镜像右位心,还需警惕是否存在先天性血管环畸形。血管环会对气管和食管产生不同程度的压迫而产生狭窄梗阻,造成呼吸困难等症状。胸部大血管增强CT及三维重建技术可以显示血管与气管、食管的位置关系;纤维支气管镜检查可以明确是否存在气管狭窄。但该患儿的胸部大血管增强CT和气道重建检查结果未见气管狭窄和异常血管环,故不支持先天性血管环畸形。
排除上述疾病,结合患儿症状及辅助检查,考虑:(1)因患儿存在内脏反位这一特点,考虑存在先天发育畸形,需明确是否为遗传性疾病或染色体异常、其他综合征等;(2)请呼吸专科医师会诊,解读纤维支气管镜检查结果,因鼻腔、气管腔内见脓性分泌物,考虑存在纤毛功能障碍,确诊需要黏膜活检电镜检查,基因检测可作为补充。结合这两点,因新生儿行黏膜活检困难,故行基因检测进一步明确。
3 进一步检查
征得家属同意后,采集患儿及其父母外周静脉血3 mL(EDTA抗凝)送检迈基诺医学检验所行一家三口全外显子基因检测,对测序结果中变异位点行Sanger测序验证。基因结果提示患儿DNAH5基因存在1个大片段杂合缺失和1个半合子突变。前者为exon 48_50杂合缺失,来源于患儿母亲。后者为c.7915C>T(p.R2639X),第48号外显子第7 915号核苷酸由胞嘧啶C变为胸腺嘧啶T(NM_001369)的半合子突变,导致氨基酸发生无义突变;患儿父亲该位点杂合变异,母亲该位点无变异(图3),人类基因突变数据库(HGMD,http://www.hgmd.cf.ac.uk/)已有该位点与Kartagener综合征的致病性报道。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)[1]遗传变异分类标准和指南,DNAH5基因exon 48_50大片段杂合缺失可能导致基因功能丧失,符合PVS1;该变异为genomAD数据库(http://gnomad.broadinstitute.org)、DGV数据库(http://dgv.tcag.ca/dgv/app/home)正常对照人群中未发现的变异,符合PM2;因此该变异被判定为可能致病性变异。DNAH5基因c.7915C>T(p.R2639X)为无义突变,可能导致基因功能丧失,符合PVS1;该变异为千人数据库正常对照人群中未发现的变异,在ESP数据库(http://evs.gs.washington.edu/EVS)、ExAC数据库 (http://exac.broadinstitute.org)正常对照人群中频率极低,符合PM2;HGMD数据库已有该位点隐性遗传的病例报道,变异标签为致病突变,ClinVar数据库(https://www.ncbi.nlm.nih.gov/clinvar)对该位点的致病性分析为致病性,与原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)相关,符合PM3;因此该变异被判定为致病性变异。
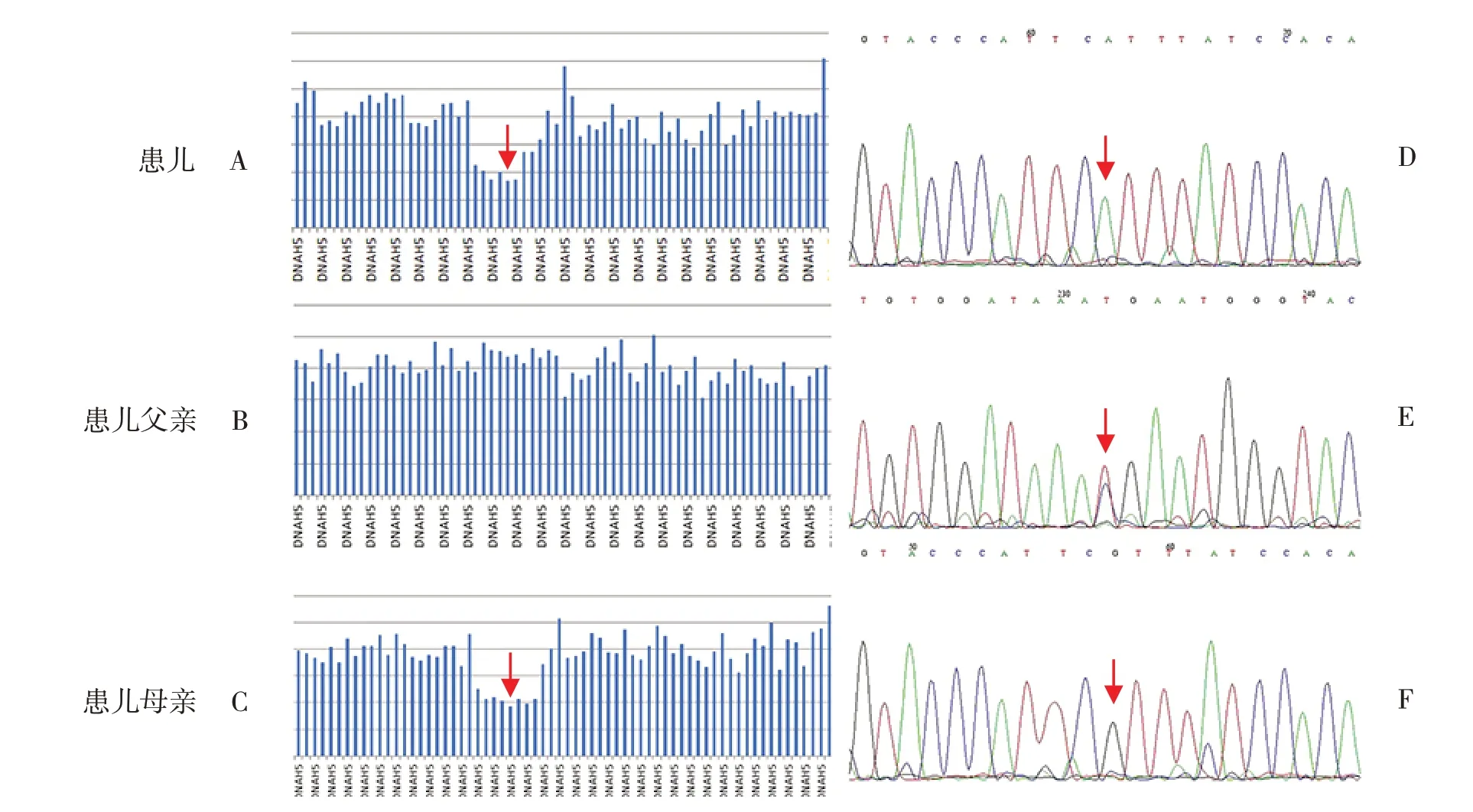
图3 患儿及其父母DNAH5基因测序图 图A~C为拷贝数变异分析结果,提示患儿DNAH5基因存在exon48_50杂合缺失(箭头所指区域),患儿父亲无缺失,患儿母亲杂合缺失。图D~F为DNAH5基因突变位点Sanger测序图(c.7915C>T),提示患儿DNAH5基因存在c.7915C>T的半合子突变,患儿父亲该位点杂合变异,母亲该位点无变异。箭头所指为突变位点。
4 临床经过
入院后予以氧疗,头罩吸氧2 d后因仍有气促,且血气分析提示PCO2进行性上升,给予改高流量鼻导管吸氧5 d,缓解后再予鼻导管吸氧22 d后停用。因CRP升高,予以青霉素、头孢噻肟抗感染治疗5 d。同时给予胸部物理治疗,吸痰,布地奈德、沙丁胺醇雾化,氨溴索化痰等支持治疗。患儿住院22 d后,病情稳定,予以出院。出院1个月门诊随访,患儿大气吸入下氧饱和度稳定,无需吸氧,暂无呼吸道感染疾病发生。
5 诊断及诊断依据
诊断:Kartagener综合征。诊断依据:(1)足月儿,生后出现气促、发绀等呼吸困难表现。(2)体检及辅助检查提示内脏反位。(3)纤维支气管镜检查提示鼻腔和支气管腔内可见脓性分泌物。(4)存在DNAH5基因有1个杂合缺失和1个半合子突变。
6 讨论
纤毛是从上皮细胞游离面伸出来,类似毛发的细胞器。传统上,纤毛分为3类:初级纤毛、节点纤毛、运动纤毛。节点纤毛在建立左-右主体定位中起着至关重要的作用,在胚胎期,节点纤毛具有引导正常胚胎心脏向左侧移动的作用,其异常可导致偏侧缺陷,包括内脏反位和一系列内脏位置不明的情况,这可能也与先天性心脏异常有关。纤毛功能障碍及内脏反位[2],即定义为Kartagener综合征,是一种纤毛结构和/或功能异常的常染色体隐性遗传病,最先由Kartagener在1933年命名,是PCD的一种,约占PCD的50%。其特点是纤毛结构异常和/或功能损害,纤毛清除黏液功能障碍。
Kartagener综合征的典型表现为支气管扩张、慢性鼻窦炎、内脏反位三联征。在不同的年龄阶段,其临床表现也不尽相同,但呼吸道症状是疾病经典表现的一部分:包括反复的肺部感染,伴发热、咳嗽、脓痰、咯血,甚至加重而伴有胸闷、气短等。但因呼吸道疾病的大多数症状或体征在健康儿童中也常见,因此对该疾病的正确诊断有可能延后[3]。在欧洲,调查表明诊断PCD时的中位年龄是5.3岁,如伴有内脏反位的患儿确诊的平均年龄是3.5岁[4]。靳雨婷等[5]调查中国55例和国外61例Kartagener综合征患儿,发现国内诊断年龄为(9.2±3.7)岁,比国外更晚。但对于新生儿来说,没有支气管扩张、鼻窦炎等典型的呼吸道症状,因此对于新生儿期诊断Kartagener综合征是难点。研究发现,超过85%以上的PCD患儿存在新生儿呼吸窘迫症状[6]。造成呼吸困难的原因并不明确,可能是因为需要把羊水从肺部清除。通常这种呼吸困难不是像新生儿常见呼吸疾病(如湿肺、新生儿呼吸窘迫综合征)在生后不久发生,而是在12~24 h后发生[7];部分患儿可能是在生后数天或数周出现呼吸困难。此外,部分患儿可能会有鼻塞、咳嗽等[8]。
因此,临床上无法解释的新生儿呼吸窘迫需要警惕PCD。研究表明超过75%的PCD足月新生儿需要持续吸氧数天至数周[9];对于出现新生儿呼吸窘迫的患儿,PCD需要更长的吸氧时间,平均需要15.2 d,明显长于对照组(0.8 d)[10]。如果新生儿出现不能解释的呼吸窘迫,同时伴有肺叶萎陷、内脏反位,和/或氧疗时间大于2 d的,需考虑PCD[11]。本例患儿气促等呼吸困难症状排除了常见疾病,治疗后仍有气促症状,通过检查进一步明确了诊断,这也是国内首例报道的在新生儿期确诊的Kartagener综合征。
Kartagener综合征患儿的胸部X线片可能会出现肺部上叶和中叶萎陷,通常会被诊断为新生儿肺炎[12]。但影像学上新生儿早期并无支气管扩张、鼻窦炎典型表现。支气管镜的检查需要呼吸专科医师操作或协助会诊,相对于新生儿医师更有经验,能进一步提高诊断率。
通过黏膜活检电镜检查纤毛的超微结构是诊断该病的金标准[13],可通过鼻刷取下鼻甲黏膜或支气管镜检查取下气道黏膜获得上皮标本。动力臂缺失是最常见的结构异常,其他包括动力臂数目减少、微管排列紊乱等[10]。除了临床症状及辅助检查结果,因新生儿黏膜活检困难,基因检测是明确诊断的重要补充手段[14]。
DNAH5和DNAI1是目前已知的PCD患者最主要的致病基因,分别在28%和10%的PCD患者中发现[15]。DNAH5基因突变通常与外动力臂缺陷、内脏转位和男性不育有关[16];DNAI1基因突变可导致外动力臂变短或缺失。除此之外,其他发现的基因突变包括:DNAH11[17],其编码外动力蛋白重链,与内脏转位有关;DNAI2可致外动力臂缺陷,因其可控制身体左右极性,突变可致身体随机的左右不对称[18];ARMC4的突变可导致外动力臂缺陷,致内脏转位和纤毛不能运动[19]。本例患儿经基因检查明确为DNAH5基因突变,支持Kartagener综合征的诊断。
目前临床上并无Kartagener综合征的特效疗法,以对症治疗为主。建议所有患者都应进行常规临床就诊,以通过痰或口咽培养进行肺活量测定监测和呼吸道培养监测[20]。在任何年龄,建议每年至少进行2~4次就诊,如果呼吸道症状恶化,则应根据培养和药敏试验结果选择合适的抗生素治疗。此外,最新的研究也表明基因治疗可能是未来的新方向,通过“基因编辑”[21]的方法可以恢复正常的基因功能和纤毛运动,从而达到治疗目的。基因治疗正在兴起一个新时代,但仍需更多的临床研究证实。
对于Kartagener综合征或PCD患儿的治疗需要团队合作,包括儿科医生、呼吸科医生、物理治疗师、耳鼻喉科医生、遗传学家、放射科医生、男科医生等,为患者提供长期的治疗和随访,各专科联合提供综合治疗方案。当然,如患者能早期诊断,及早接受治疗和管理会获得更好的临床结局,预后相对良好[22]。
7 结语
新生儿期出现呼吸困难及合并内脏反位,如无法用常见疾病解释时,应当考虑到Kartagener综合征。黏膜活检电镜检查是目前诊断该病的金标准,对于新生儿或小婴儿活检困难者,基因检测可作为明确诊断的补充条件;及时识别和正确处理是本病的临床难点。本文总结1例以新生儿期出现呼吸困难、合并内脏反位为主要表现的Kartagener综合征的诊疗过程,以提高临床医师对本病的认识,减少漏诊、误诊,及早发现,纳入长期管理以获得相对良好的预后。
猜你喜欢
自然杂志(2022年3期)2022-08-18
医学研究生学报(2021年4期)2021-12-02
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
散文诗世界(2019年6期)2019-09-10
中国临床医学影像杂志(2019年6期)2019-08-27
意林·全彩Color(2019年7期)2019-08-13
中华皮肤科杂志(2019年5期)2019-06-24
百科知识(2015年18期)2015-09-10
西南医科大学学报(2015年1期)2015-08-22
