
湖北地区茯苓饮片质量标准研究
2021-06-10 05:30:28王运芳林华清陈剑锋
华中师范大学学报(自然科学版) 2021年3期
王运芳,秦 文,林华清,陈剑锋,邹 坤*
(1.天然产物研究与利用湖北省重点实验室,三峡大学生物与制药学院,湖北 宜昌 443002;2.湖北恒安芙林药业有限公司,湖北 宜昌 443103)
茯苓Poriacocos(Schw.) Wolf 为多孔菌科卧孔菌属真菌茯苓的干燥菌核[1-2],主要分布在我国云南、湖北、安徽、湖南等地[3],是中国延用千年的传统中药,可与多种中药相互配伍,且不分季节,不管寒、温、风、湿等疾病,都能发挥其独特功效,亦可食用[4].经研究,茯苓化学成分有三萜类、多糖类、甾体、脂肪酸、蛋白质、腺嘌呤、组氨酸、树胶、胆碱、卵磷脂、氨基酸以及钙、镁、铁、钾等无机元素.其中主要成分为多糖和三萜类成分[5],茯苓多糖属于真菌多糖,主要由水溶性多糖和碱溶性多糖组成[6].在三萜类化合物中去氢茯苓酸、茯苓酸、松苓新酸等含量较高[7-11].研究表明其有调节免疫功能、抗肿瘤、抗炎等多方面药理作用[12-13],还有保肝、抗衰老、降血糖等作用[14-15].
主要受药材的产地、采收时间、加工方法和贮存等因素的影响,茯苓饮片的质量参差不齐.本课题参照国家药监局发布的《中药配方颗粒质量控制与标准制定技术要求》,在中医药理论的指导下对茯苓饮片进行了系统的研究,建立了茯苓中水溶性多糖的含量测定方法,HPLC法测定茯苓中茯苓酸、去氢茯苓酸和松苓新酸的含量,在《中国药典》(2020年版)的基础上丰富了茯苓饮片质量标准的内容.为提高茯苓药材的利用率,促进中药材的产地深加工和产业结构调整,并带动中药材种植业、中医药知识经济等相关产业的发展提供了较为科学的依据.
1 材料与仪器
1.1 仪器
Ultimate 3000 型高效液相色谱仪(戴安公司);电子天平(上海民桥精密仪器有限公司);SX-2.5-10型马弗炉(上海洪纪仪器设备有限公司);UV-3100Spectrophotometer(上海美谱达仪器有限公司);BCD-228CH冰箱(河南新飞电器集团有限公司);电子天平(上海民桥精密仪器有限公司);78-1型磁力加热搅拌器(常州国华电器有限公司);SHZ-D(Ⅲ)循环水式多用真空泵(郑州科丰仪器设备有限公司);DLSB-5/-20℃ 低温冷却液循环泵(巩义市予华仪器有限责任公司);N-1100 旋转蒸发仪(上海爱朗仪器有限公司);SB-2000 水浴锅(上海爱朗仪器有限公司);GZX-9240 MBE 数显鼓风干燥箱(上海博迅实业有限公司医疗设备厂);低温离心机(德国 Eppendorf公司).
1.2 试剂
石油醚(60~90 ℃)(国药集团化学试剂有限公司);正丁醇(国药集团化学试剂有限公司);三氯甲烷(国药集团化学试剂有限公司);乙酸乙酯(天津市富宇精细化工有限公司);95%乙醇(国药集团化学试剂有限公司);浓硫酸(河南信阳化工厂);重蒸苯酚(北京鼎国昌盛生物技术有限公司);甲醇(TEDIA公司);乙腈(TEDIA公司);纯净水(华润怡宝饮料(中国)有限公司).甲醇和乙腈为色谱纯,其他试剂均为分析纯.
1.3 药材
茯苓饮片10个批次(批号161101~161110)的原料经三峡大学医学院曾建红教授鉴定为多孔菌科卧孔菌属真菌茯苓Poriacocos(Schw.) Wolf,植物标本现保存于天然产物研究与利用湖北省重点实验室.饮片的样品信息见表1.
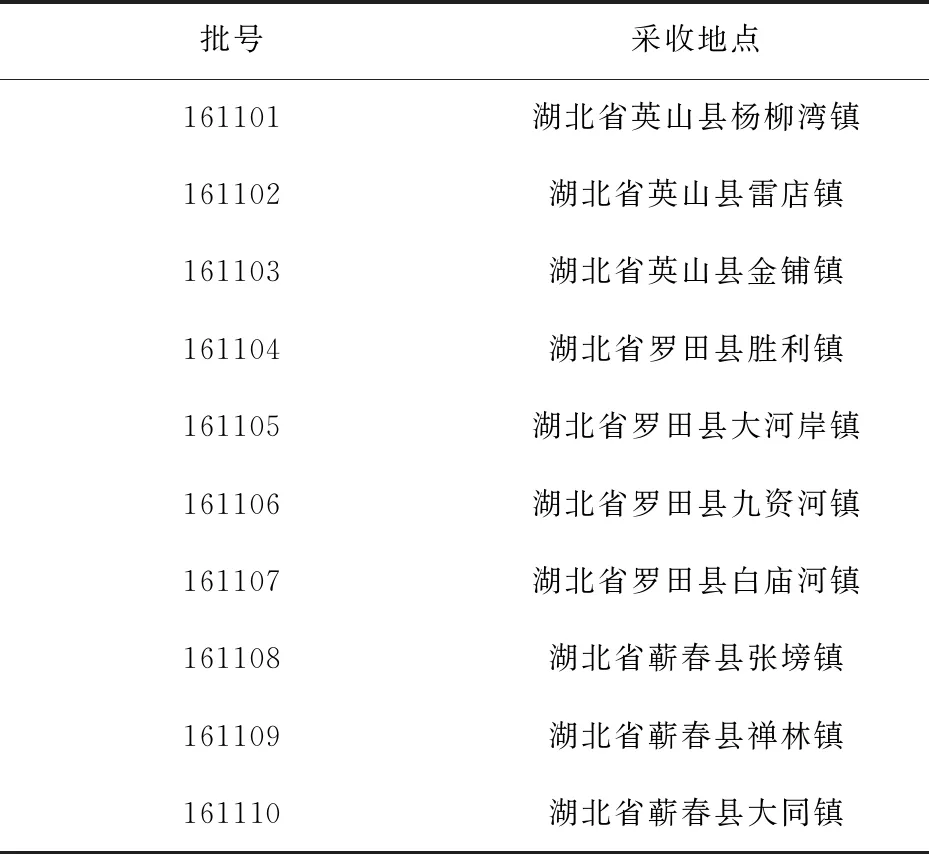
表1 茯苓饮片的样品来源信息Tab.1 Sources of processed slices of Poria cocos
2 方法与结果
2.1 形状和颜色
茯苓饮片共10个批次,均为去皮后切制成的立方块状,可大小不一,颜色白色或淡棕色.
2.2 鉴别
主要参照《中国药典》2020年版一部收载茯苓饮片项下的检验方法.由图片可见菌丝无色或淡棕色,细长,稍弯曲,有分枝,直径3~8 μm,少数至16 μm.见图1(放大200倍).
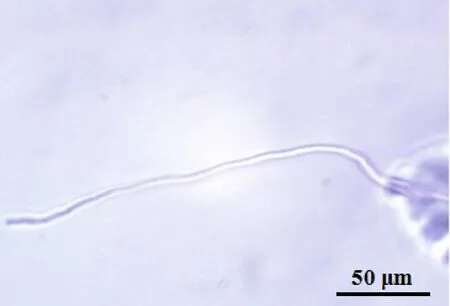
图1 茯苓饮片的显微鉴定图Fig.1 Microscopic identification of decoction piece of Poria cocos
2.3 水分的测定
精密称取茯苓饮片粉末(过 100目筛)各5 g,按《中国药典》2020年版通则0832第二法(烘干法),分别测定10批次茯苓饮片的含水量,结果见表2.依据《中国药典》2020年版,水分不得超过18.0%,故10批次样品中有6批次不合格.

表2 茯苓饮片水分含量测定结果(n=3)Tab.2 Determination results of water content in decoction slices of Poria cocos (n=3)
2.4 总灰分的测定
精密称取茯苓饮片粉末(过 100目筛)各5 g,按《中国药典》2020年版通则2302,分别测定10批次茯苓饮片的总灰分,结果见表3.仅有一批次样品总灰分超过2.0%为不合格.

表3 茯苓饮片总灰分含量测定结果(n=3)Tab.3 Determination results of total ash content in decoction slices of Poria cocos (n=3)
2.5 浸出物的测定
精密称取茯苓饮片粉末(过100目筛)各5 g,按照《中国药典》2020年版通则2201醇溶性浸出物测定法项下的热浸法测定,结果见表4.按照2020版标准,醇溶性浸出物不得少于2.5%,则10批样品均合格.
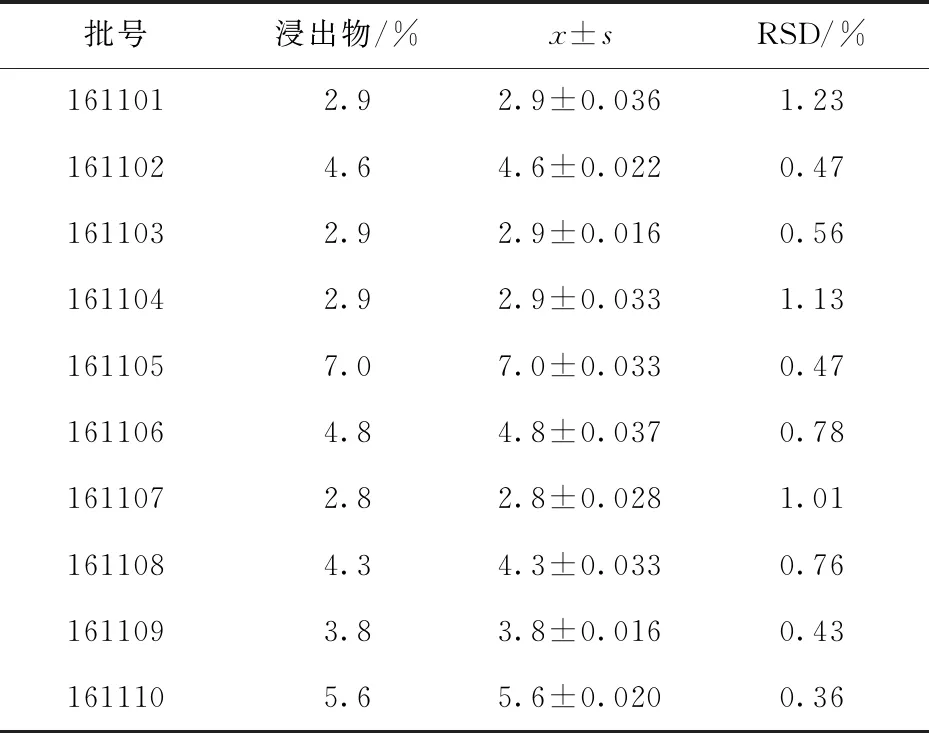
表4 茯苓饮片浸出物的测定结果(n=3)Tab.4 Determination results of extract from decoction slice of Poria cocos (n=3)
2.6 茯苓饮片二氧化硫残留量
精密称取茯苓饮片粉末(过 100目筛)各10 g,按照《中国药典》2020年版通则2331二氧化硫残留量测定法项下的第一法(酸碱滴定法),结果见表5.
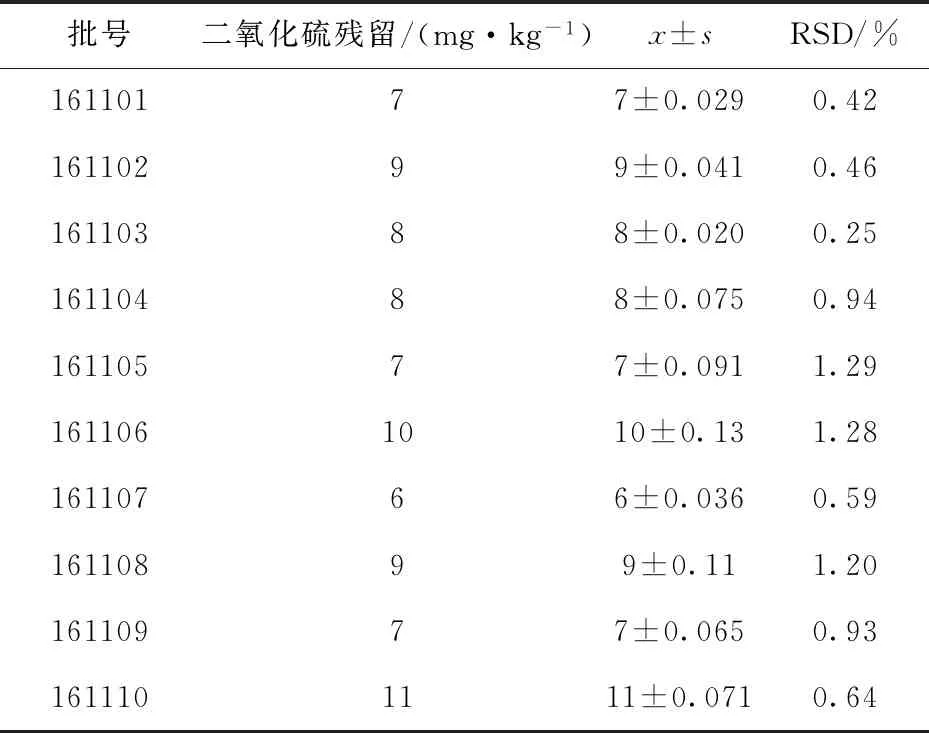
表5 茯苓饮片二氧化硫残留量的测定结果(n=3)Tab.5 Determination results of sulfur dioxide residue in decoction slices of Poria cocos (n=3)
由表5可知,10批次茯苓饮片的二氧化硫残留量在7~11 mg·kg-1范围内,为茯苓饮片质量标准制定提供参考依据.
2.7 茯苓饮片中水溶性多糖的含量测定
茯苓粗多糖和精制多糖的制备[16]:称取茯苓粉末10 g(过60目筛,50 ℃烘干24 h),置索氏提取器中,加入石油醚100 mL,90℃回流提取2 h,抽滤.药渣挥尽有机溶剂后,置于圆底烧瓶中,加蒸馏水100 mL,称总重量,水浴回流2 h后擦干烧瓶外表面,称重后加蒸馏水补足减失重量,摇匀,抽滤.精密量取续滤液50 mL,浓缩至5 mL.加入95%乙醇25 mL,于4℃冰箱中放置过夜,离心(3 500 r·min-1,15 min).弃去上清液,将沉淀干燥至恒重,即得到茯苓粗多糖.
将茯苓粗多糖用5 mL蒸馏水复溶,加5 mL Sevage试剂(氯仿∶正丁醇=4∶1),混匀后反复振摇、离心(2 000 r·min-1,10 min),留下水层,重复多次去除蛋白.将水层先在200 ~ 400 nm紫外光谱段进行扫描,260 ~ 280 nm范围内无吸收峰,再用考马斯亮蓝法测蛋白含量.向浓缩液中加入适量30% H2O2原液(体积比),用0.5 mol·L-1的NaOH溶液调节其pH至8,45 ℃水浴4 h,将多糖脱色液置于透析袋中,透析袋中无气泡,两端需夹紧.先用自来水流动透析1 d,再用蒸馏水透析3 d,每12 h更换一次蒸馏水.透析后过滤,浓缩至5 mL.加入95%乙醇25 mL,于4 ℃冰箱中放置过夜,离心(3 500 r·min-1,15 min),弃去上清液,将沉淀依次用无水乙醇、丙酮、乙醚洗涤3次,50 ℃真空干燥,即得茯苓精制多糖.
2.8 PMP衍生化法测定粗多糖和精制多糖的单糖组成
衍生化供试品的制备:精密称取茯苓粗多糖10 mg和茯苓精制多糖5 mg,吹氮气,各加入2 mol·L-1三氟乙酸1 mL(通风处取用),摇匀,立即封管,水浴6 h.从中各取出400 μL水解液置具塞试管中,加甲醇适量,氮气吹干,重复操作直到完全清除三氟乙酸.在水解供试品中加0.3 mol·L-1NaOH溶液和0.3 mol·L-1的PMP溶液各100 μL,混合均匀,于70 ℃水浴100 min,冷却至室温后加0.3 mol·L-1HCl溶液100 μL中和,加蒸馏水至1 mL,最后各加入1 mL三氯甲烷除去未反应的PMP,弃去油相,重复3次.将茯苓粗多糖和茯苓精制多糖水相分别以0.45 μm滤膜过滤后,取续滤液备用.
HPLC条件:色谱柱型号为依利特C18柱,250 mm × 4.6 mm,5 μm;流动相为乙腈(A)-0.1 mol·L-1磷酸盐缓冲液(pH6.7)(B),等梯度洗脱,有机相和水相比例为18(A):82(B);二极管阵列UV-VIS检测器;检测波长为250 nm;柱温为25 ℃,流速为1 mL·min-1;进样量为20 μL.多糖标准品、茯苓粗多糖、茯苓精制多糖的PMP衍生物HPLC图分别见图2(a)、2(b)和2(c).
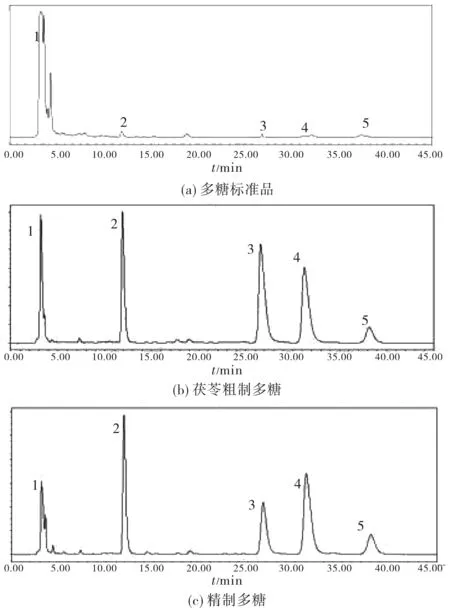
1.甘露糖mannitose;2.鼠李糖rhamnose;3.葡萄糖glucose;4.半乳糖galactose;5.木糖xylose图2 多糖标准品、茯苓粗多糖和精多糖的PMP衍生物HPLC图Fig.2 HPLC chromatograms of PMP derivatives of standard polysaccharides,crude polysaccharides of Poria cocos and refined polysaccharides
由图2可知,茯苓多糖精制前后多糖中甘露糖、鼠李糖、葡萄糖、半乳糖和木糖的组成没有发生明显变化.
供试品溶液的制备.称取茯苓粉末10 g(过60目筛,50 ℃烘干24 h),置索氏提取器中,加入石油醚100 mL,90 ℃回流提取2 h,抽滤.药渣挥尽有机溶剂后,置于圆底烧瓶中,加蒸馏水100 mL,称总重量,水浴回流2 h后擦干烧瓶外表面,称重后加蒸馏水补足减失重量,摇匀,抽滤.精密量取续滤液50 mL,浓缩至5 mL.加入95%乙醇25 mL,于4 ℃冰箱中放置过夜,离心(3 500 r·min-1,15 min).分离上清液,备用.将沉淀用蒸馏水溶解,定容至100 mL容量瓶中,精密移取2.0 mL至10.0 mL容量瓶中,蒸馏水定容至刻度,摇匀,即得水溶性多糖供试品溶液.
对照品溶液的制备.精密称量干燥至恒重的葡萄糖对照品0.100 4 g,加蒸馏水定容至100 mL,摇匀,制成浓度为1.004 mg·mL-1的储备液.精密移取此溶液10 mL于50 mL棕色容量瓶中,蒸馏水定容至刻度,摇匀,即得.
最大吸收波长的选择.精密吸取葡萄糖对照品溶液和供试品溶液各1 mL,分别置20 mL 试管中,分别精密加入现配的5%重蒸苯酚溶液1 mL,振摇均匀后,立即精密移取浓硫酸6 mL,贴壁缓慢加入,边加边振摇均匀,避光静置40 min后摇匀.在紫外可见分光光度仪上,于200 ~ 700 nm进行光谱扫描,选吸收值最大的波长.对照品溶液和供试品溶液最大吸收波长都在489 nm.
标准曲线的绘制.分别精密吸取对照品溶液适量,配制成10.040 μg·mL-1、20.080 μg·mL-1、40.120 μg·mL-1、60.240 μg·mL-1、80.320 μg·mL-1、120.480 μg·mL-1的系列溶液.精密移取以上葡萄糖对照品溶液各1.0 mL,于20 mL试管中,按照“2.12”项下显色方法显色并测定吸光度.经计算得回归方程:y=0.0076x+0.0864(r=0.9995,n=5),葡萄糖在10.040 ~ 120.480 μg·mL-1范围内线性关系良好.
转换因子的测定精密称取2.5 mg茯苓精制多糖于EP管中,加蒸馏水溶解后转移到50 mL棕色容量瓶,并将EP管洗涤3次,洗涤液全部转移到到容量瓶,加蒸馏水至刻度线定容,摇匀.精密移取1 mL茯苓精制多糖溶液于具塞试管中,按照“2.12”项下显色方法显色并测定吸光度,根据回归方程计算多糖溶液中葡萄糖的浓度,根据公式
F=m/c×D,
其中,m为多糖质量(mg),c为多糖中葡萄糖浓度(mg·mL-1),D为稀释倍数,测得换算因子F=2.10 (n=5).
2.9 精密度试验
精密量取1 mL葡萄糖对照品溶液,按“2.12”项下方法显色并测定其吸光度,平行测定6次,记录吸光度值,根据回归方程计算对照品溶液的葡萄糖含量.计算结果RSD=0.46%,表明此台紫外可见分光光度计的精密度良好.
2.10 稳定性试验
按照“2.7”项下提取方法制备1份茯苓供试品溶液(批号161101).精密量取供试品溶液1 mL于具塞试管中,按照“2.12”项下方法显色并测定吸光度值1,静置1 h后摇匀,测定并记录吸光度值2;静置2 h后摇匀,测定并记录吸光度值3;静置2 h后摇匀,测定并记录吸光度值4.根据回归方程计算0~3 h内4个时间点的供试品溶液的浓度,并计算结果的RSD=0.59%,表明供试品溶液在3 h内显色稳定.
2.11 重复性试验
按照“2.7”项下提取方法平行制备6份茯苓供试品溶液(批号161101).精密量取各供试品溶液1 mL,分别置于具塞试管中,按照“2.12”项下方法显色并测定吸光度,结果RSD=3.01%表明该方法重复性良好.
2.12 样品含量测定
精密量取各批次茯苓水溶性多糖供试品溶液各1.0 mL,精密加入1.0 mL现配的5%重蒸苯酚溶液,充分混合后,缓慢精密加入6.0 mL浓硫酸,振摇均匀后,立即精密移取浓硫酸6.0 mL,贴壁缓慢加入,边加边振摇均匀,避光静置40 min后摇匀.于紫外可见分光光度仪上,以吸收波长489 nm测定样品溶液的吸光度值,并根据回归方程计算供试品中水溶性多糖的含量.结果见表6.
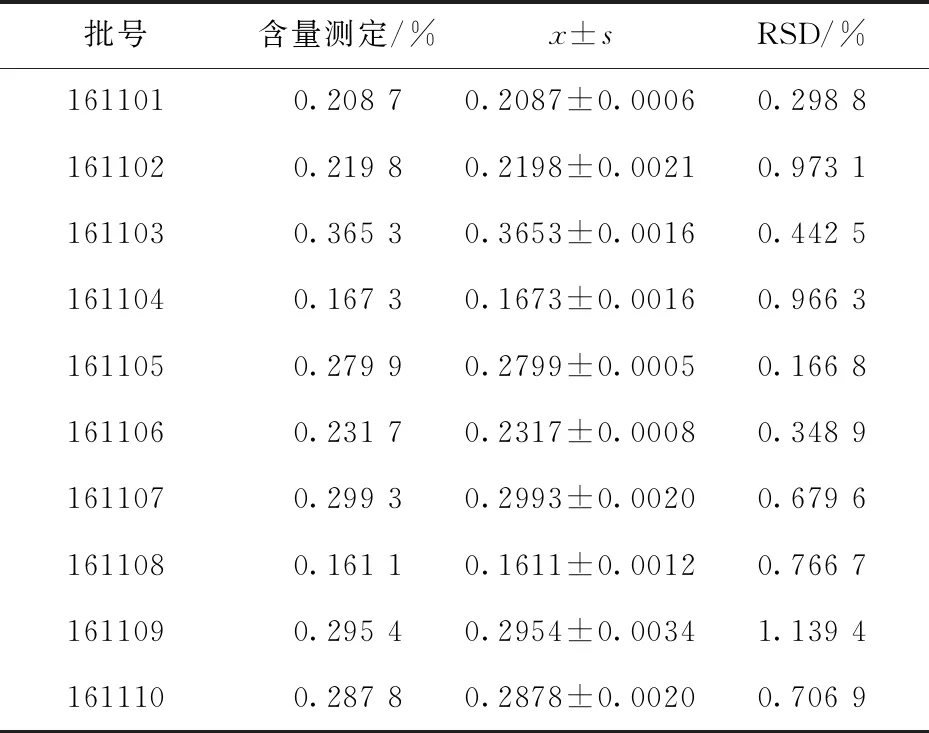
表6 茯苓供试品中水溶性多糖的含量测定结果(n=3)Tab.6 content determination results of water-soluble polysaccharide in Poria Cocos samples(n=3)
2.13 加样回收率试验
称取已知含量的同一批茯苓样品粉末(批号161110) 6 份,每份约0.2 g,分别加入精制多糖约22 mg,按照“2.7”项下制备供试品溶液和“2.12”项下方法显色并测定吸光度,计算回收率,结果平均回收率为99.14%,RSD=1.01%,表明方法准确度良好,结果见表7.
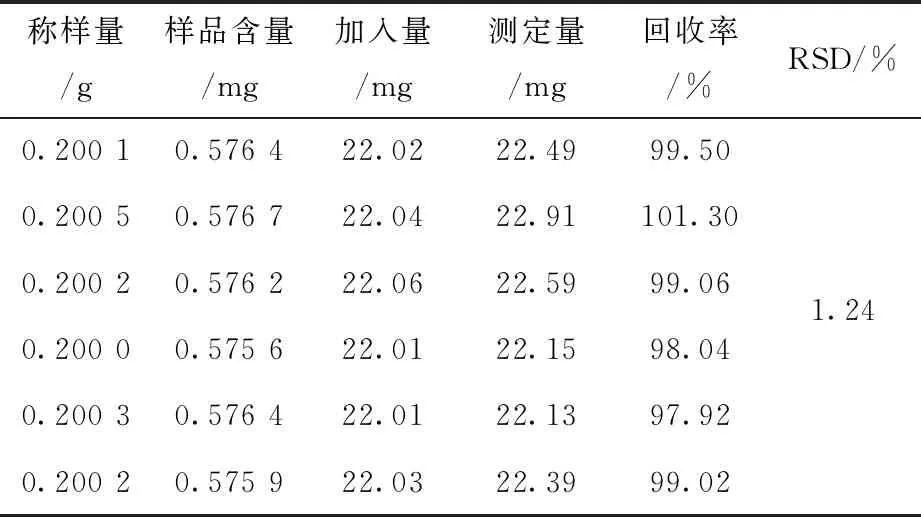
表7 茯苓多糖加样回收率实验(n=6)Tab.7 Recovery of Poria Cocos Polysaccharide (n=6)
3 茯苓饮片中茯苓酸、去氢茯苓酸和松苓新酸的含量测定
3.1 供试品及对照品溶液的配制
茯苓酸对照品溶液:精密称取4.583 mg茯苓酸对照品于10 mL容量瓶中,加适量甲醇溶解,并定容,摇匀.即得浓度为458.3 μg·mL-1的对照品溶液,备用.
去氢茯苓酸对照品溶液:精密称取1.001 mg去氢茯苓酸对照品于5 mL容量瓶中,加适量甲醇溶解,并定容,摇匀.即得浓度为200.2 μg·mL-1的储备液,备用.
松苓新酸对照品溶液:精密称取1.834 mg松苓新酸对照品于5 mL容量瓶中,加适量甲醇溶解,并定容,摇匀.即得浓度为366.7 μg·mL-1的储备液,备用.
供试品溶液的配制:取“2.7”项下分离的上清液,50 ℃减压浓缩后冷冻干燥至恒重.精密称量样品各0.1 g,用甲醇定容至5.0 mL容量瓶,摇匀,0.45 μm滤膜过滤,取续滤液,即得供试品溶液,备用.
HPLC条件色谱柱:依利特C18( 250 mm×4.6 mm,5 μm);检测波长:去氢茯苓酸和松苓新酸为254 nm,茯苓酸为210 nm;流速:1 mL·min-1;进样量:50 μL;温度:25 ℃.流动相条件,液相条件为:0~30 min为甲醇:0.25%磷酸水溶液=10:90~65:35;30~90min为甲醇∶0.25%磷酸水溶液=65∶35~90∶10;90~95 min为甲醇∶0.25%磷酸水溶液=90∶10.
3.2 专属性试验
在选定的条件下,茯苓酸、去氢茯苓酸和松苓新酸对照品与样品中其它组分色谱峰可基线分离,无干扰.按茯苓酸、去氢茯苓酸和松苓新酸峰计算,理论塔板数为4 000以上.茯苓酸、去氢茯苓酸和松苓新酸对照品、供试品的高效液相色谱图见图3.
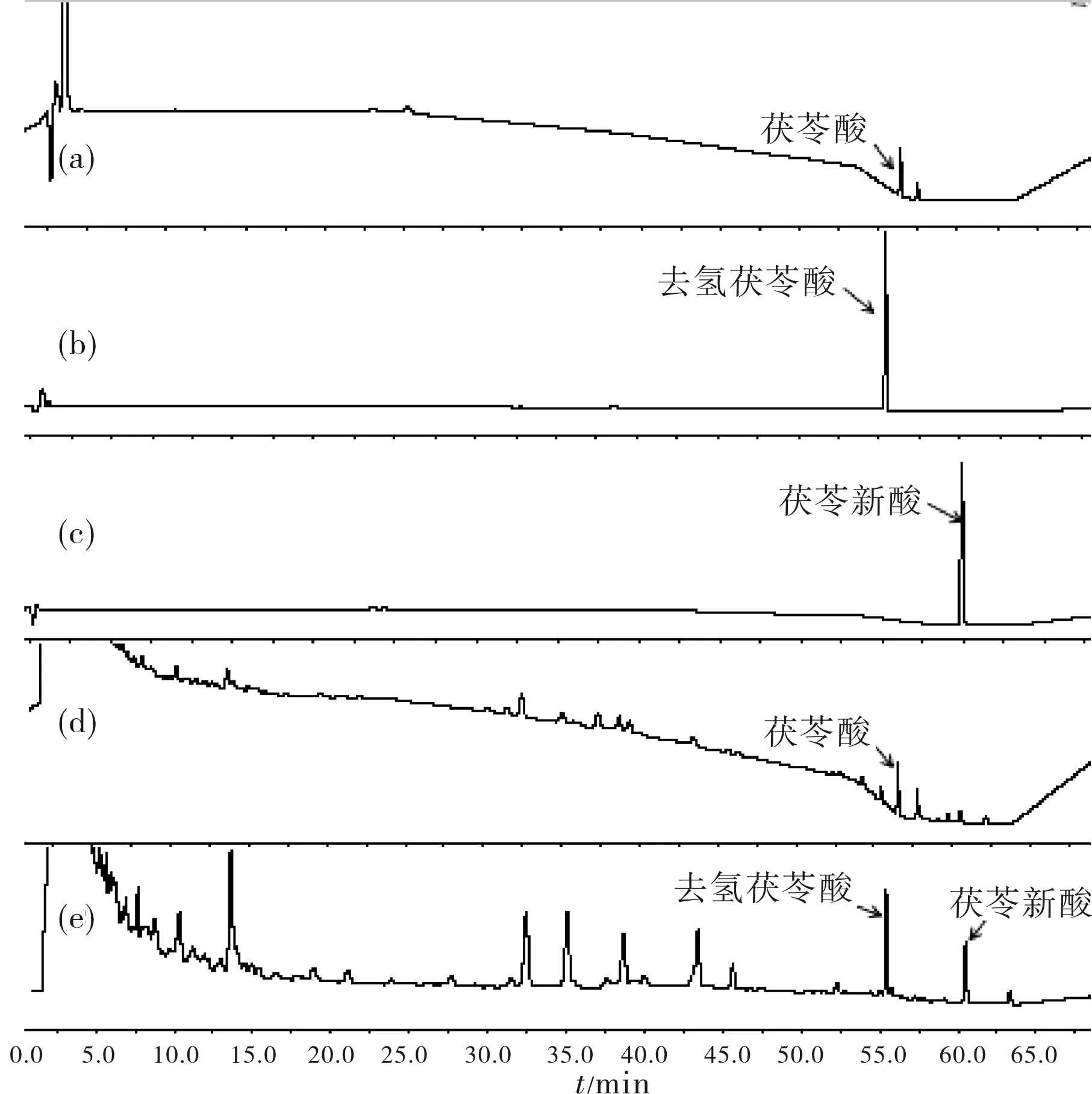
(a)茯苓酸(210 nm);(b)去氢茯苓酸(254 nm);(c)松苓新酸(254 nm);(d)茯苓样品(210 nm);(e)茯苓样品(254 nm)(a)Pachymicacid (210 nm);(b)Dehydropachymic acid (254 nm);(c)Poricoic acid (254nm);(d)Poria cocos sample (210 nm);(e) Poria cocos sample (254 nm)图3 对照品和样品HPLC图谱Fig.3 HPLC chromatogram of reference substance and sample
3.3 线性关系的考察
分别精密吸取系列梯度浓度的茯苓酸、去氢茯苓酸、松苓新酸对照品溶液10 μL不同浓度注入液相色谱仪中,按“3.1”项下HPLC条件测定其峰面积.茯苓酸、去氢茯苓酸、松苓新酸的线性范围如表8所示.

表8 茯苓酸、去氢茯苓酸和松苓新酸的线性范围Tab.8 Linear ranges of porylic acid,dehydrogenated porylic acid and pinoleic acid
结果表明茯苓酸、去氢茯苓酸、松苓新酸分别在在1.4~458.3 μg·mL-1、4.2~250.0 μg·mL-1及0.6~91.7 μg·mL-1范围内呈良好线性关系.
3.4 精密度试验
分别精密吸取对茯苓酸、去氢茯苓酸、松苓新酸对照品溶液10 μL,重复进样3次,计算得出茯苓酸、去氢茯苓酸、松苓新酸的RSD分别为0.61%、1.56%和1.69%(n=3),结果表明仪器精密度好.
3.5 稳定性试验
精密吸取同一供试品溶液,分别于0、1、2、4、8、24 h各进样10 μL,以茯苓酸、去氢茯苓酸、松苓新酸峰面积计算,RSD分别为l.l4%、1.68%和1.95%(n=3),结果表明供试品溶液在24 h内稳定.
3.6 重复性试验
精密称定同一批次茯苓饮片10 g,共3份,按上述方法制备供试液,进样测定,计算得茯苓酸、去氢茯苓酸、松苓新酸的RSD分别为1.90%、1.37%和1.12%(n=3),结果表明该法重复性好.
3.7 样品含量测定
取“3.1”项下供试品溶液,按“3.1”项下HPLC条件测定其峰面积.计算得样品中茯苓酸、去氢茯苓酸和松苓新酸的平均百分含量分别为0.269%、0.593%和0.379%.详细百分含量结果见表9.
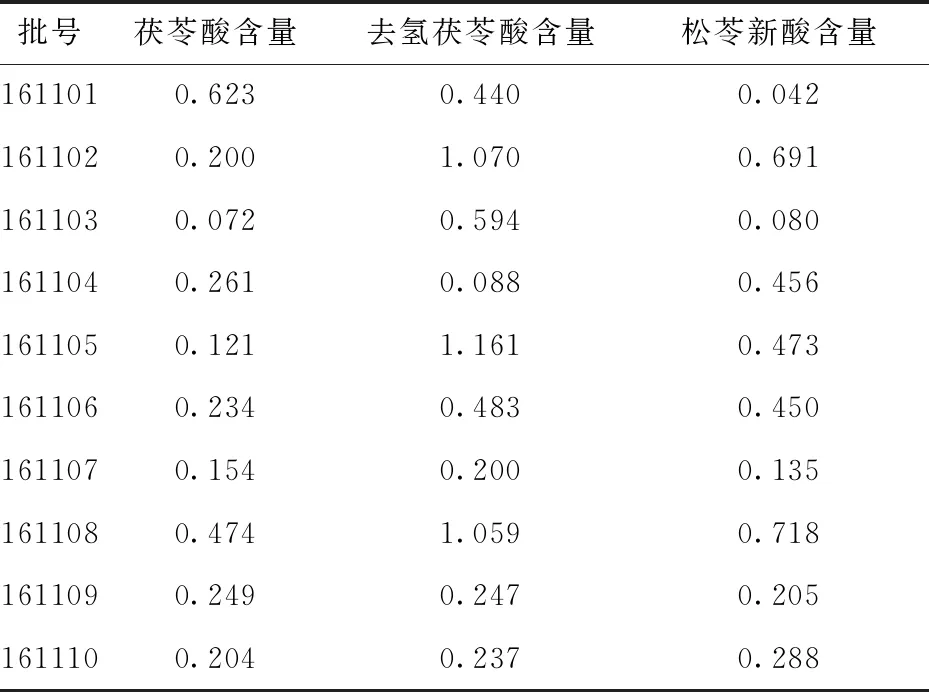
表9 茯苓饮片中茯苓酸、去氢茯苓酸和松苓新酸的含量测定(n=3)Tab.9 Determination of content of poria acid, dehydrogenated poria acid and pinoleic acid in prepared slices of poria cocos(n=3) /%
3.8 加样回收率试验
取已知含量的同一批次茯苓样品粉末(161110)约0.05 g,于10 mL容量瓶中,加适量甲醇溶解,平行3份.另取茯苓酸对照品、去氢茯苓酸对照品及茯苓新酸对照品,将3种对照品分别用甲醇制成每1 mL 含0.150 2 mg、0.150 1 mg及0.150 3 mg的混合对照品溶液.精密加入上述混合对照品溶液1 mL ,并用甲醇定容,按供试品溶液制备方法操作.按“3.1”项下方法HPLC条件进行测试,计算回收率,茯苓酸的平均回收率为100.3%(RSD=1.70%),去氢茯苓酸平均回收率为101.3%(RSD=1.94%),松苓新酸平均回收率为100.9%(RSD=1.45%)表明方法准确度良好.
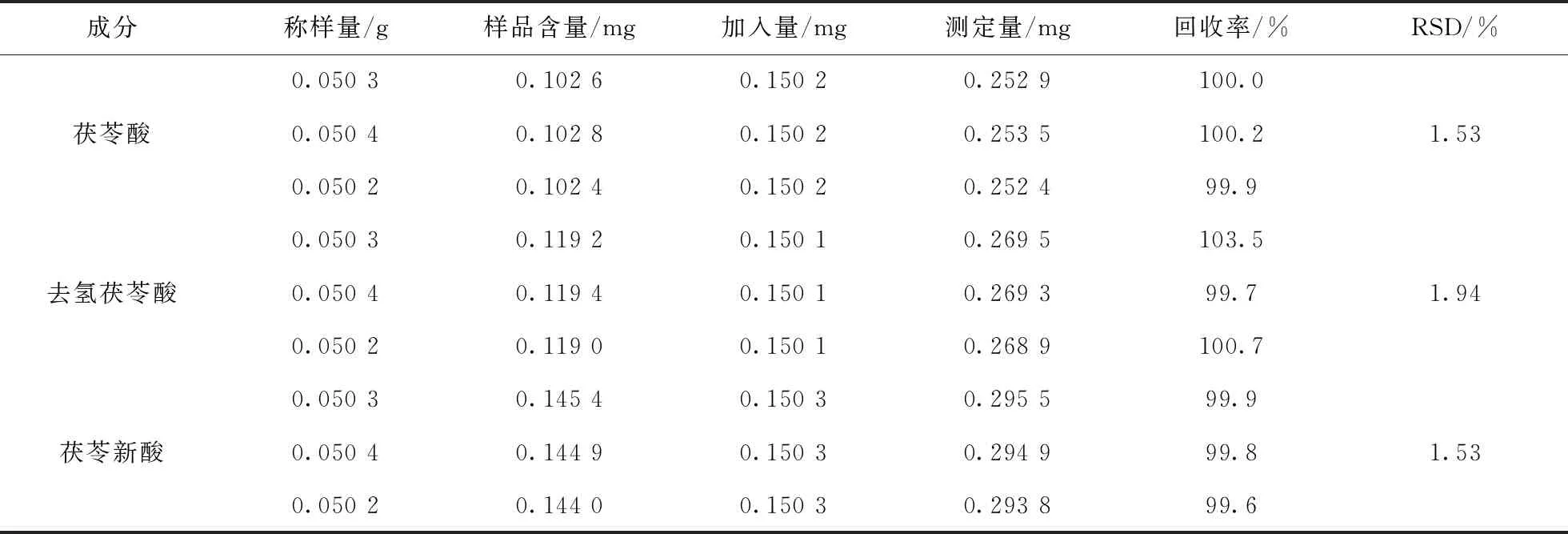
表10 茯苓酸、去氢茯苓酸、松苓新酸加样回收率实验(n=3)Tab.10 The recovery rateswith pachylic acid,dehydropachylic acid and pachylic neoacid were added (n=3)
4 讨论
《中国药典》2020年版茯苓饮片质量标准中规定了性状、鉴别、检查、浸出物等项的检验,但这些指标不能全面、系统的控制饮片的质量.因茯苓在加工、贮藏等环节仍可能存在随意使用硫磺熏蒸的情况,故在本文中增加了二氧化硫残留量的研究;且药材质量的优劣在一定程度上可以通过有效成分含量的高低来反映,因此对茯苓中的多糖及三种三萜类化合物即茯苓酸、去氢茯苓酸和松苓新酸进行了进一步研究.
经测定10批次茯苓饮片含水量为15.6%~30.5%;总灰分为0.2%~2.8%;醇溶性浸出物为2.9%~7.0%;10批次茯苓饮片中均能不同程度地检出二氧化硫,范围为6~11 mg·kg-1;水溶性多糖的平均百分含量为0.251 6%.茯苓饮片中茯苓酸、去氢茯苓酸、松苓新酸的平均百分含量分别为0.259 2%、0.557 9%、0.353 8%,且别在1.4~458.3 μg·mL-1、4.2~250.0 μg·mL-1和0.6~91.7 μg·mL-1范围内呈良好线性关系.按照《中国药典》2020年版规定分析出10批次药品中水分检测项中不合格的有6批,总灰分检测项中不合格的有1批;溶性浸出物检测中均合格;10批次样品中均能检测出二氧化硫.综上所述,本文对茯苓饮片质量标准进行了较为系统的研究,该方法简便、稳定、专属性强、重复性好,且本课题可提高茯苓药材的利用率,促进中药材的产地深加工和产业结构调整,并带动中药材种植业、中医药知识经济等相关产业的发展.
猜你喜欢
Journal of Traditional Chinese Medicine(2022年5期)2022-10-14 11:38:16
世界科学技术-中医药现代化(2021年5期)2021-11-05 06:55:18
基层中医药(2020年7期)2020-09-11 06:38:04
中成药(2019年12期)2020-01-04 02:02:50
Advances in Petroleum Exploration and Development(2019年1期)2019-08-05 09:23:32
基层中医药(2018年10期)2018-12-06 09:27:20
中成药(2018年3期)2018-05-07 13:34:17
机电信息(2014年35期)2014-02-27 15:54:29
机电信息(2014年26期)2014-02-27 15:53:34
中国现代中药(2012年6期)2012-10-30 01:38:26
