
肠炎沙门氏菌攻毒对SPF雏鸡粪便微生物区系的影响
2021-06-08 01:26王曦
家禽科学 2021年3期
王曦
摘 要:试验旨在探究肠炎沙门氏菌攻毒对SPF雏鸡粪便微生物区系的影响。选择1日龄SPF雏鸡24只,随机分为2组,每组12只。在4日龄处理组口服107 CFU肠炎沙门氏菌(ATCC13076)攻毒,对照组口服等量PBS溶液。试验期22 d。结果显示:①处理组SPF鸡的存活率为75%,对照组全部存活,期间两组鸡的体重无显著性差异;②OTU水平上,相同日龄两组鸡粪便菌群的丰富度相近,中期处理组粪便菌群多样性更高(P<0.05),后期对照组菌群稳定性更高,而且攻毒对SPF鸡粪便菌群的影响随日龄增加而减弱;③在属水平上,与对照组相比,处理组Bacteroides(拟杆菌门)、Enterococcus(肠球菌属)和Aeriscardovia(卡多维亚氏菌属)的相对丰度明显升高(P<0.05);Lactobacillus(乳杆菌属)、Alistipes(另枝菌属)、Faecalibacterium(粪杆菌属)、Romboutsia(罗姆布茨菌属)和Ruminococcaceae_UCG_005(瘤胃球菌UCG_005)的相对丰度明显降低(P<0.05);④KEGG分析显示攻毒影响了菌群参与的多个代谢通路。综上,肠炎沙门氏菌感染影响了SPF雏鸡粪便菌群的结构组成、稳定性及菌群参与的代谢通路。Bacteroides(拟杆菌门)、Lactobacillus(乳杆菌属)等差异菌属可能对SPF雏鸡抵抗沙门氏菌入侵起到积极作用。
关键词:肠炎沙门氏菌;SPF雏鸡;16S rRNA测序;粪便菌群;KEGG代谢通路
动物肠道中含有极其复杂的共生微生物群组分,其抵抗病原菌入侵产生的定殖抗性对宿主的健康起到保护作用[1]。肠道微生物的组成会随着时间的推移而发展[2],也会随着病原菌入侵而发生变化[3]。肠炎沙门氏菌是一种常见人畜共患病的肠道病原体,引起宿主肠道炎症反应,临床表现一般为腹泻。在腹泻症状消失后,肠炎沙门氏菌可以在宿主体内隐性携带,并伴随着长期的间歇性排菌[4]。在刚孵化的SPF鸡肠道菌群单一的窗口期引入肠炎沙门氏菌,使其更容易引起肠道菌群结构的变化[5]。而我们感兴趣的是早期肠炎沙门氏菌感染对SPF雏鸡粪便菌群结构产生怎样的影响。本研究利用16S rRNA基因高通量测序技术来探究肠炎沙门氏菌攻毒对SPF雏鸡粪便菌群的影响,并结合日龄来研究粪便菌群的相关性变化。
1 材料和方法
1.1 试验设计与试验动物 试验选取山东省农业科学院家禽研究所SPF鸡胚场孵化的1日龄SPF雏鸡24只,随机分成两组,每组12只,其中处理组在4日龄时口服0.2 mL(约107CFU)的肠炎沙门氏菌(ATCC13076),对照组口服等量无菌PBS溶液,分别在隔离器内隔离饲养,正常饲喂雏鸡饲料和饮水。
1.2 生存曲线、体重与粪便样品采集 每天观察鸡的生长情况,实验结束后绘制生存曲线。在第1、4、5、7、9、11、14、18和22日龄时称量每组鸡的体重。在第5、7、9、18和22日龄收集两组SPF鸡的粪便,每组随机采集6份新鲜粪便,每份都收集到一个5 mL离心管里,加入少量PBS缓冲液混匀后,-20℃保存,用于检测粪便菌群多样性。
1.3 粪便菌群16S rRNA高通量测序及分析
1.3.1 DNA提取和PCR扩增 根据E.Z.N.A.soil试剂盒(Omega Bio-tek, Norcross, GA, U.S.) 说明书进行总DNA抽提,DNA浓度和纯度利用NanoDrop 2000进行检测,利用1%琼脂糖凝胶电泳检测DNA提取质量,用338F(5'- ACTCCTACGGGAGGCAGCAG-3')和806R (5'- GGACTACHVGGGTWTCTAAT-3')引物对 V3~V4可变区进行PCR扩增,扩增程序为:95 ℃预变性3 min,27个循环(95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s),最后72 ℃延伸10 min(PCR仪:ABIGeneAmp?9700型)。扩增体系为20 μL,4 μL 5×Fast Pfu缓冲液、2 μL 2.5 mMdNTPs、0.8 μL引物(5 μM)、0.4 μLFastPfu聚合酶和10 ngDNA模板。
1.3.2 Illumina Miseq测序 使用2%琼脂糖凝胶回收PCR产物,利用AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA)進行纯化,Tris-HCl洗脱,2%琼脂糖电泳检测。利用QuantiFluorTM-ST(Promega, USA) 进行检测定量。根据Illumina MiSeq平台 (Illumina,San Diego, USA)标准操作规程将纯化后的扩增片段构建PE 2×300的文库。利用Illumina公司的Miseq PE300平台进行测序(上海美吉生物医药科技有限公司)。
1.4 数据处理与分析 测序序列使用 Trimmomatic软件质控,使用FLASH软件进行拼接:①设置50 bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截去后端碱基,去除质控后长度低于50 bp的序列;②barcode需精确匹配,引物允许2个碱基的错配,去除模糊碱基;③根据重叠碱基overlap将两端序列进行拼接,overlap需大于10 bp,去除无法拼接的序列。使用的UPARSE 软件(version 7.1 http://drive5.com/uparse/),根据97%的相似度对序列进行OTU聚类;使用UCHIME软件剔除嵌合体。利用RDP classifier(http://rdp.cme.msu.edu/)对每条序列进行物种分类注释,比对Silva数据库(SSU123),设置比对阈值为70%。主坐标分析(Principal coordinates analysis,PCoA)、线性判别分析(Linear discriminant analysis Effect Size,LEfSe)、冗余分析(Redundancy analysis,RDA)和PICRUSt功能预测分析在上海美吉生物免费云平台进行(https://www.i-sanger.com/)。变异系数(coefficient of variation, CV)为标准差与平均值的商(SD/Mean)。在软件GraphPad Prism7中利用Mann-Whitney 算法进行差异显著性检验。
2 结果和分析
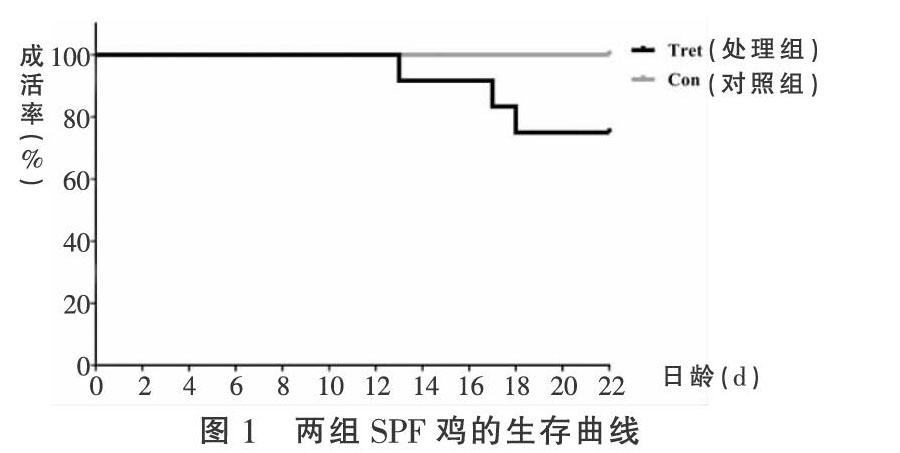
2.1 生存曲线与体重变化 对照组存活率为100%,处理组存活率为75%,见图1。在14~22日龄,处理组SPF雏鸡的均重都大于对照组,但没有显著性差异(P>0.05),见表1。
2.2 16S rRNA基因高通量测序数据分析
2.2.1 OTU水平上粪便菌群结构与差异分析 在方法与材料中已经列出了两组SPF鸡5个时间点的粪便样本。在进行Illumina MiSeq测序和质量过滤后,为了降低每个粪便样本间的测序深度差异造成的影响,这些序列被统一抽平至样本中最小序列数(每个样本28423个序列)。OTU水平上,两组在5个日龄除了重叠的OTU,各自都含有差异OTU,见表2。
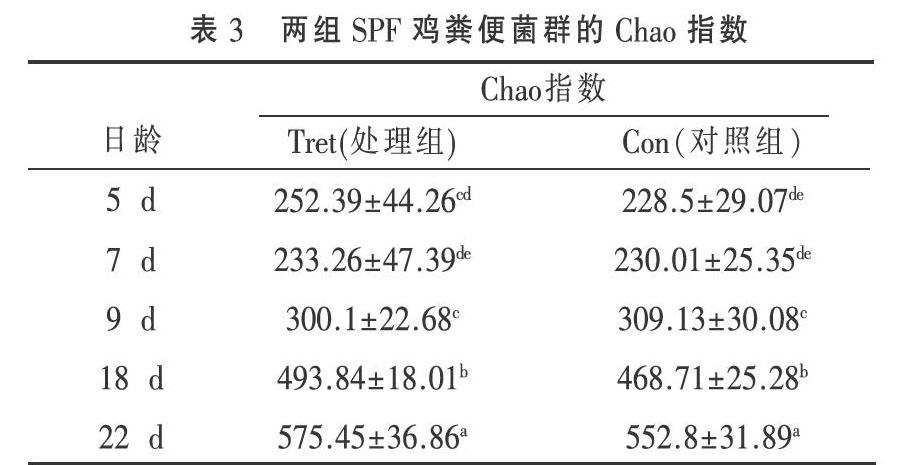
通过α多样性分析比较了两组粪便样本的Chao指数和Shannon指数,发现比较相同日龄处理组与对照组粪便菌群Chao指数始终无显著性差异,见表3;而在第5、9、18日龄时处理组的Shannon指数显著大于对照组,见表4。
Chao和Shannon指数变异系数用来衡量SPF鸡粪便菌群的稳定性。结果发现,对照组粪便菌群的稳定性大于处理组,见图2。
基于Brays-Curtis 算法的主坐标分析样本的β多样性。根据组别和年龄所有样本均可以分为10个不同的小组,在PCoA图上对日龄的分析显示出样品的异质分布,随着时间的推移,两组的群落结构愈加相似,见图3A;另一方面,考虑到攻毒的PCoA分析表明,在每个采样时间点,处理组和对照组的细菌群落结构明显不同,见图3B。
2.2.2 门、属级粪便菌群结构组成与差异分析 在门分类水平上,两组的菌门(相对丰度>1%)都由Firmicutes(厚壁菌门)、Bacteroidetes(拟杆菌门)、Actinobacteria(放线菌门)和Proteobacteria(变形菌门)组成,见图4A和4B。
Actinobacteria与Proteobacteria的平均相对丰度在两组均处于较低水平。两组中Firmicutes的相对丰度均在18日龄之前超过95%,从第18日龄起,两组Firmicutes的相對丰度开始下降的同时,伴随着Bacteroidetes的相对丰度迅速上升。18~22日龄,处理组的Firmicutes平均相对丰度极显著低于对照组(P<0.01),处理组的Bacteroidetes平均相对丰度极显著高于对照组(P<0.01),见图5。
在属分类水平,有24个属的细菌相对丰度在两组样本中曾大于1%,在第7日龄之前,两组粪便菌群都比较单一,第9日龄之后菌群多样性迅速上升。两组的粪便菌群主要由Lactobacillus(乳杆菌属)细菌组成,处理组粪便菌群含有更多的Bacteroides(拟杆菌属)和Enterococcus(肠球菌属)细,而对照组Alistipes、Romboutsia和Faecalibacterium等菌属更加丰富。在第5~18日龄,Lactobacillus都是最丰富的属,丰度随时间推移而缓缓降低。第22日龄,处理组中Bacteroides相对丰度最高,约25.8%;在处理组中Alistipes相对丰度最高,约18.5%,结果见图4C。
使用LEfSe分析比较了在同一日龄两组间的差异菌群。在第5和7日龄无差异菌属(阈值≥4),所以只呈现了第9、18、22日龄的LDA得分。我们发现在这3个日龄中共出现了8个差异菌属(见图6A~C),处理组处于优势水平的有3个菌属,分别是Enterococcus、Aeriscardovia和Bacteroides。对照组处于优势水平的有5个菌属,分别是Romboutsia、Lactobacillus、Ruminococcaceae_UCG_005、Faecali-bacterium和Alistipes。基于LDA得分进行RDA分析,表明肠炎沙门氏菌攻毒影响了SPF雏鸡肠道菌群结构,且肠炎沙门氏菌攻毒与Ruminococcaceae_UCG_005、Romboutsia、Faecali-bacterium、Alistipes和Lactobacillus成负相关,与Enterococcus、Bacteroides和Aeriscardovia成正相关,见图6D。
通过LEfSe分析得出了差异菌属在不同日龄的丰度变化。处理组中Enterococcus在9日龄之后持续处于较高丰度水平且显著高于对照组;Aeriscardovia的丰度在9~18日龄显著高于对照组,Bacteroides的丰度在18日龄之后显著高于对照组;处理组中Romboutsia和Lactobacillus的丰度在多数日龄显著小于对照组,Ruminococcaceae_UCG_005、Alistipes和Faecalibacterium的丰度在22日龄显著小于对照组,见图7。
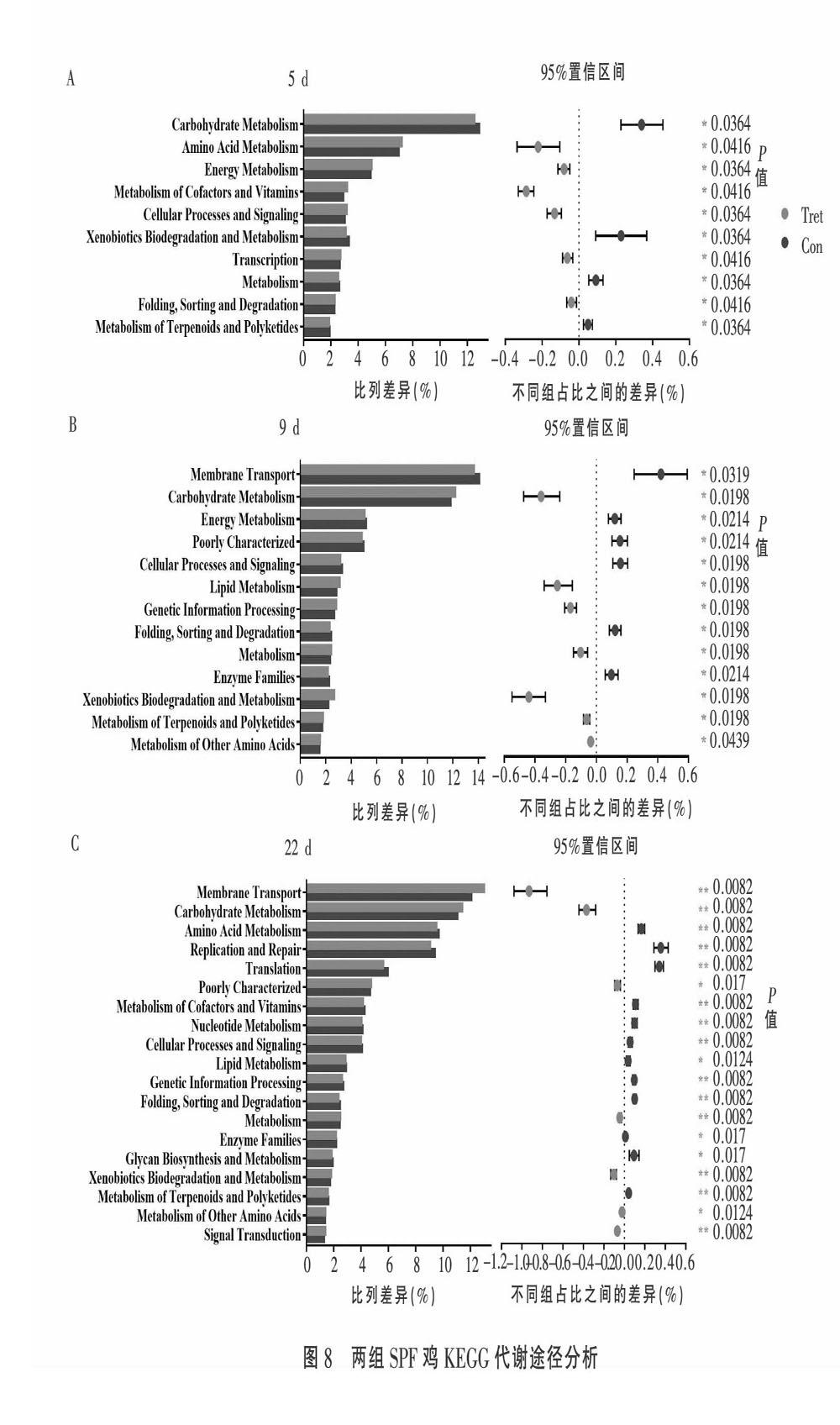
2.2.3 KEGG代谢途径差异分析 经与KEGG数据库比对,发现在第5、9和22日龄有显著差异的代谢通路共有22个,其中3个日龄共同的有6个;第5和22日龄共同的有2个;第9和22日龄共同的有6个。攻毒后,从日龄发展上看,处理组中代谢能力(Metabolism)、外源生物降解与代谢(Xenobiotics Biodegradation and Metabolism)、碳水化合物代谢(Carbohydrate Metabolism)、不良特征(Poorly Characterized)、其他氨基酸代谢(Metabolism of Other Amino Acids)和膜运输(Membrane Transport)水平升高,且后期显著高于对照组(P<0.05);处理组中折叠、分类和降解(Folding, Sorting and Degradation)、细胞过程和信号(Cellular Processes and Signaling)、辅助因子和维生素的代谢(Metabolism of Cofactors and Vitamins)、氨基酸代谢(Amino Acid Metabolism)、遗传信息处理(Genetic Information Processing)和脂质代谢(Lipid Metabolism)水平下降,且后期显著低于对照组(P<0.05)。有6条代谢通路在第22日龄时第一次出现,其中信号转导(Signal Transduction)在处理组显著高于对照组(P<0.01);酶家族(Enzyme Families)、聚糖生物合成与代谢(Glycan Biosynthesis and Metabolism)、核苷酸代谢(Nucleotide Metabolism)、翻译(Translation)、复制和修复(Replication and Repair)在处理组显著低于对照组(P<0.05),见图8。
3 讨论
16S rRNA基因高通量测序分析得出两组鸡粪便菌群丰富度和多样性都随着日龄的增加而增加,与已报道的结果一致[2],且粪便菌群的丰富度在相同日龄没有显著差异,但处理组的菌群多样性在9~18日龄显著高于对照组,说明攻毒导致了SPF鸡粪便菌群多样性增加[6],这或许与处理组SPF鸡对抗肠炎沙门氏菌的入侵相关。在22日龄时,对照组粪便菌群稳定性均高于处理组,说明攻毒导致了SPF鸡粪便菌群稳定性变差。同时处理组多样性的增加也表明了多样性不能成为肠道菌群好坏的判定标准[7]。两组菌群的β多样性分析使用了基于Brays-Curtis算法的PCoA,5个日龄中处理组与对照组的粪便菌群OTU明显分开。总体的样本分布表明,随着时间的推移,两组的菌群结构愈加接近,攻毒对SPF鸡的粪便菌群结构的影响随时间推移逐渐变弱。
两组鸡粪便菌群在门水平(相对丰度>1%)组成一致,都由Firmicutes、Bacteroidetes、Actinobacteria和Proteobacteria组成[8];其中Firmicutes始终为最丰富的菌门[9]。第18和22日龄,Bacteroidetes的相对丰度急剧攀升,且处理组中Bacteroidetes的相对丰度显著高于对照组(P<0.01)。总体来看,Firmicutes与Bacteroidetes是两组SPF鸡粪便菌群中比重最大的菌门[9],攻毒使处理组的菌门波动比对照组更明显。
肠炎沙门氏菌入侵后会劫持宿主吞噬体系统,为避免被宿主清除而形成含沙门氏菌液泡(Salmonella-containing vacuoles,SCVs),并从其中招募膜和蛋白质。SCV通过与胞吞和循环途径的持续相互作用而成熟,从成熟的SCV延伸出来的广泛的管膜网络,促进了宿主细胞信号、膜运输和一些蛋白质的生成[10]。
在第18日龄之后,Bacteroides在处理组的相对丰度显著高于对照组(P<0.01),它具有增强鸡新陈代谢的能力,分解难消化碳水化合物(包括纤维素和淀粉)促进碳水化合物代谢,合成丙酸[11-13]。丙酸对沙门氏菌具有抑制作用[14]。处理组中Enterococcus在9日龄之后均保持在较高的丰度水平,它作为一种重要的家禽病原体,可以通过产生细胞外超氧化物和过氧化氢来损伤结肠上皮细胞中的真核细胞DNA[15],引起的局部细胞损伤和活性氧可能改变肠道通透性,导致细胞死亡,从而触发炎症和肥胖等不良表征[16]。处理组中外源生物降解与代谢的增强应该是由肠炎沙门氏菌攻毒引起的,这或许可以作为动物状态异常的标志。
在18日龄之前,Lactobacillus的相对丰度在两组中一直处于很高的水平(两组相对丰度均大于0.5);第22日龄时,处理组Lactobacillus的相对丰度显著低于对照组(P<0.01)。Lactobacillus可合成乳酸来调节肠道pH值,可发酵低聚糖(低聚果糖和低聚葡萄糖)、多糖(菊粉),促进聚糖生物合成与代谢,刺激产丁酸盐的细菌生长[17-19]。第22日龄时,Ruminococcaceae_UCG-005、Alistipes和Faecalibacterium在处理组中的相对丰度显著低于对照组(P<0.01)。Ruminococcaceae_UCG-005在消化多糖和纤维方面起着重要作用[20]。
Ruminococcaceae被认为是潜在的有益菌,具有抗炎活性,参与了肠道环境的正向调节,并与免疫调节和健康的动态平衡有关[21]。Lactobacillus和Alistipes都可以将色氨酸代谢成吲哚[22, 23],吲哚的存在降低体内沙门氏菌侵袭,可与短链脂肪酸(丙酸、丁酸)的协同增强抗沙门氏菌的作用,降低沙门氏菌毒力基因(SPI-1)表达,增强上皮细胞对沙门氏菌侵袭的抵抗力[24],吲哚可减轻宿主细胞炎症并增加肠上皮细胞屏障的完整性[25]。Faecalibacterium属是一种功能高度活跃的有益菌,它消耗醋酸和产生大量的丁酸,Sokol等人在人体外细胞模型和小鼠体内结肠炎模型中发现Faecalibacterium均具有不依赖丁酸额外的抗炎特性[26]。因此Faecalibacterium对肠道稳态至关重要,在促进肠道健康方面具有潜在的重要作用。丁酸盐可以提高脂肪利用率,增强脂肪代谢[27],减少沙门氏菌感染[28],并能刺激上皮细胞生长[29]。第9日龄后,Romboutsia在对照组的相对丰度显著高于处理组(P<0.01)。Romboutsia可利用来自宿主的L-岩藻糖[30],而宿主能够调节其肠道上皮细胞的岩藻糖基化反应,以岩藻糖为能源的微生物可能有助于保护宿主免受内源性病原体的感染[31]。虽然目前没有充分的功能性研究证明Romboutsia可以促进宿主健康,但它依然具有成为益生菌的潜力。
健康的微生物区系对于宿主消化道有着重要作用,如:产生各种酶,促进消化道发育和肠粘膜增殖,维生素合成和利用,发酵和内源产品的利用等[32]。处理组的粪便菌群中脂质代谢、聚糖生物合成与代谢、酶家族、细胞过程和信号、氨基酸代谢、折叠、分类和降解、辅助因子和维生素的代谢、遗传信息处理、核苷酸代谢、翻译、复制与修复等代谢水平降低,可能是因为肠炎沙门氏菌的入侵损伤了菌群结构的结果。以上这些代谢通路的变化可在一定程度上解释肠炎沙门氏菌入侵与粪便微生物相对丰度的变化相关关系。
4 结论
随着日龄的推移,粪便菌群的丰富度与多样性增加的同时也变得更加稳定。肠炎沙门氏菌感染影响了SPF鸡早期粪便菌群的结构组成、稳定性以及菌群参与的代谢通路,处理组中占优的Bacteroides以及对照组占优的Lactobacillus、Alistipes、Faecalibacterium和Ruminococcaceae_UCG_005等有益菌可能对SPF鸡抵抗沙门氏菌入侵起到積极作用。未来需要进一步深入研究这些显著变化的差异菌在调控肠道菌群中的具体作用及机制,为将来临床开发利用某种益生菌来改善动物表现奠定基础。
參考文献:
[1] LIBERTUCCI J, YOUNG V B. The role of the microbiota in infectious diseases[J]. Nature microbiology, 2019,4(1):35-45.
[2] CHAMBERS J R, GONG J. The intestinal microbiota and its modulation for Salmonella control in chickens[J].Food research international, 2011,44(10):3149-3159.
[3] LUPP C, ROBERTSON M L, WICKHAM M E, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae[J]. Cell host & microbe, 2007,2(2):119-129.
[4] CARTER A J,ADAMS M R,WOODWARD M J,et al.Control strategies for Salmonella colonisation of poultry:the probiotic perspective[J]. Food science & technology bulletin: functional foods, 2009,5(9):103-115.
[5] ROTO S M,KWON Y M,RICKE S C. Applications of In Ovo Technique for the Optimal Development of the Gastrointestinal Tract and the Potential Influence on the Establishment of Its Microbiome in Poultry[J].Frontiers in veterinary science, 2016,3:63.
[6] VAN DONGEN W F, WHITE J, BRANDL H B, et al.Age-related differences in the cloacal microbiota of a wild bird species[J]. BMC ecology, 2013,13:11.
[7] FOSTER K R, SCHLUTER J, COYTE K Z, et al. The evolution of the host microbiome as an ecosystem on a leash[J].Nature,2017,548(7665):43-51.
[8] WEI S, MORRISON M, YU Z. Bacterial census of poultry intestinal microbiome[J]. Poultry science, 2013,92(3):671-683.
[9] MANCABELLI L, FERRARIO C, MILANI C, et al. Insights into the biodiversity of the gut microbiota of broiler chickens[J]. Environmental microbiology, 2016,18(12):4727-4738.
[10] VORWERK S, KRIEGER V, DEIWICK J, et al. Proteomes of host cell membranes modified by intracellular activities of Salmonella enterica[J]. Molecular & cellular proteomics,2015,14(1):81-92.
[11] PANDIT R J, HINSU A T, PATEL N V, et al. Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing[J]. Microbiome,2018,6(1):115.
[12] BECKMANN L, SIMON O, VAHJEN W. Isolation and identification of mixed linked beta-glucan degrading bacteria in the intestine of broiler chickens and partial characterization of respective 1,3-1,4-beta-glucanase activities[J]. Journal of basic microbiology, 2006,46(3):175-185.
[13] FISCHBACH M A, SONNENBURG J L. Eating for two:how metabolism establishes interspecies interactions in the gut[J]. Cell host & microbe, 2011,10(4):336-347.
[14] VERMEULEN K, VERSPREET J, COURTIN C M,et al.Reduced particle size wheat bran is butyrogenic and lowers Salmonella colonization, when added to poultry feed[J].Veterinary microbiology, 2017,198:64-71.
[15] JONES B V,BEGLEY M,HILL C,et al. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome[J]. Proceedings of the National Academy of Sciences of the United States of America, 2008,105(36):13580-13585.
[16] HOU Q,KWOK L Y,ZHENG Y,et al. Differential fecal microbiota are retained in broiler chicken lines divergently selected for fatness traits[J]. Scientific reports, 2016,6:37376.
[17] HATTI-KAUL R, CHEN L, DISHISHA T, et al. Lactic acid bacteria: from starter cultures to producers of chemicals[J]. FEMS microbiology letters, 2018,365(20).
[18] YADAV M, VERMA M K, CHAUHAN N S. A review of metabolic potential of human gut microbiome in human nutrition[J]. Archives of microbiology, 2018,200(2):203-217.
[19] MEIMANDIPOUR A, SHUHAIMI M, HAIR-BEJO M, et al. In vitro fermentation of broiler cecal content: the role of lactobacilli and pH value on the composition of microbiota and end products fermentation[J].Letters in applied microbiology, 2009,49(4):415-420.
[20] ZHANG J, SHI H, WANG Y, et al. Effect of Limit-Fed Diets with Different Forage to Concentrate Ratios on Fecal Bacterial and Archaeal Community Composition in Holstein Heifers[J]. Frontiers in microbiology, 2018,9:976.
[21] SHANG Q, SHAN X, CAI C, et al. Dietary fucoidan modulates the gut microbiota in mice by increasing the abundance of Lactobacillus and Ruminococcaceae[J]. Food & function, 2016,7(7):3224-3232.
[22] ZELANTE T, IANNITTI R G, CUNHA C, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22[J]. Immunity, 2013,39(2):372-385.
[23] TYRRELL K L, WARREN Y A, CITRON D M, et al. Re-assessment of phenotypic identifications of Bacteroides putredinis to Alistipes species using molecular methods[J]. Anaerobe, 2011,17(3):130-134.
[24] KOHLI N, CRISP Z, RIORDAN R, et al. The microbiota metabolite indole inhibits Salmonella virulence:Involvement of the PhoPQ two-component system[J].PloS one,2018,13(1):e190613.
[25] BANSAL T, ALANIZ R C, WOOD T K, et al. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation[J].Proceedings of the National Academy of Sciences of the United States of America, 2010,107(1):228-233.
[26] SOKOL H, PIGNEUR B, WATTERLOT L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients[J]. Proceedings of the National Academy of Sciences of the United States of America, 2008,105(43):16731-16736.
[27] KASAHARA K, KRAUTKRAMER K A, ORG E, et al. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model[J]. Nature microbiology, 2018,3(12):1461-1471.
[28] VAN IMMERSEEL F, BOYEN F, GANTOIS I, et al.Supplementation of coated butyric acid in the feed reduces colonization and shedding of Salmonella in poultry[J]. Poultry science, 2005,84(12):1851-1856.
[29] KIEN C L, BLAUWIEKEL R, BUNN J Y, et al. Cecal infusion of butyrate increases intestinal cell proliferation in piglets[J].The Journal of nutrition, 2007,137(4):916-922.
[30] GERRITSEN J, FUENTES S, GRIEVINK W, et al.Characterization of Romboutsia ilealis gen. nov., sp. nov., isolated from the gastro-intestinal tract of a rat, and proposal for the reclassification of five closely related members of the genus Clostridium into the genera Romboutsia gen. nov.,Intestinibacter gen.nov.,Terrisporobacter gen.nov.and Asaccharospora gen. nov[J].International journal of systematic and evolutionary microbiology, 2014,64(Pt 5):1600-1616.
[31] PICKARD J M, MAURICE C F, KINNEBREW M A, et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness[J]. Nature, 2014,514(7524):638-641.
[32] LAN Y, VERSTEGEN M W A, TAMMINGA S, et al. The role of the commensal gut microbial community in broiler chickens[J]. World's poultry science journal, 2005,61(1).
猜你喜欢
健康体检与管理(2022年2期)2022-04-15
家庭医药·快乐养生(2020年11期)2020-12-03
环球时报(2020-07-28)2020-07-28
家禽科学(2020年3期)2020-05-13
祝您健康(2019年12期)2019-12-12
大众科学·下旬(2019年6期)2019-09-10
学习与科普(2019年24期)2019-09-10
农民致富之友(2019年36期)2019-01-13
国外畜牧学·猪与禽(2018年4期)2018-05-14
食品与生活(2015年12期)2015-12-14
