
超声波辅助衍生-液相色谱串联质谱法快速测定克氏原鳌虾中的硝基呋喃类代谢物残留
2021-06-04 12:35朱晓玲刘杰江丰韩智刘迪范志勇张莉王会霞
现代食品科技 2021年5期
朱晓玲,刘杰,江丰,韩智,刘迪,范志勇,张莉,王会霞
(湖北省食品质量安全监督检验研究院,湖北省食品质量安全检测工程技术研究中心,湖北武汉 430070)
硝基呋喃类药物是一类具有5-硝基-2-呋喃结构的合成类抗感染药物[1],在临床上主要作为广谱抗菌药使用,目前已知的最常用的硝基呋喃类药物主要有呋喃唑酮、呋喃它酮,呋喃西林,呋喃妥因4种(结构式见图1),该类药物曾在养殖业中被应用于预防和治疗由沙门氏菌和埃希氏菌引起的猪、牛、水产以及蜜蜂的胃肠道疾病[2]。但后来的研究发现,硝基呋喃类药物具有遗传毒性和致癌性[3],因此世界各国先后禁止该类药物使用,并规定用作食品的动物组织均不得检出硝基呋喃类药物残留,我国农业部于2002年颁布禁止使用硝基呋喃类抗生素的禁令。但在实际生产实践中,该类药物以抗菌效果好、抗菌谱宽、价格低廉且不易产生耐药性等优点,使得该类药物在畜禽养殖业和水产养殖业中仍然存在违法使用的可能性,另外,从近几年的食品安全监管中也证实,虽然该类药物早在2002年就已被列为禁用药物,但目前仍存在非法使用的情况[4],给人类健康造成威胁。
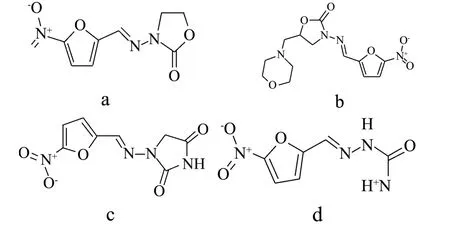
图1 4种常用的硝基呋喃类药物结构式Fig.1 Structural formula of four commonly used nitrofurans
硝基呋喃类药物进入动物体内后迅速代谢,无法直接检测动物组织中的原型药物,药物代谢后形成相应的代谢产物,分别为AMOZ、AOZ、SEM、AHD,代谢产物与动物机体蛋白质紧密结合形成稳定的化合物,因此,世界各国均以检测硝基呋喃类代谢物作为判定是否存在违法使用该类禁用药物的依据。目前,报道的硝基呋喃类药物检测方法主要有酶联免疫法[5]、极谱法[6]、拉曼光谱法[7,8]、毛细管电泳法[9]、伏安法[10]、分光光度法[11]、高效液相色谱法[12,13]和液质联用法(LC-MS/MS)[14-17]等,其中液相色谱-质谱联用法因具有高灵敏度而被广泛应用,我国现行的检测国家标准方法GB/T 21311-2007[18],农业部783号公告-1-2006[19]等,均采用的液相色谱-质谱联用法,为我国的食品安全监管提供了技术保障,但目前的标准方法以及大多数文献报道的检测方法,操作过程均较为耗时,样品的水解和衍生时间较长,需16 h,前处理复杂,而超声波辅助衍生能有效加快样品中目标化合物的水解速率和衍生反应速率,大大缩短样品前处理时间,提高检测效率,因此,开发超声波辅助衍生-液相色谱串联质谱法检测样品中的硝基呋喃类代谢物残留在实际应用中具有一定的意义。
克氏原鳌虾作为一种甲壳类水产品,多国研究报道指出[20,21],动物甲壳中可能存在非呋喃西林药物来源的氨基脲,导致食品安全监管过程中采用氨基脲进行呋喃西林药物违法使用的判定依据存在一定的争议,可能存在假阳性结果判定,造成经济损失、产业危机和消费者恐慌,因此,研究者们目前仍在持续开展带壳动物性产品中硝基呋喃类药物残留标志物或形成机制等相关研究[22]。本文以克氏原鳌虾为检测对象,建立了超声波辅助衍生,LC-MS/MS测定样品中4种硝基呋喃代谢物的分析方法,本方法改进后将衍生时间缩短到60 min,大大提高检测效率,为顺利开展后续研究奠定基础。
1 材料与方法
1.1 材料与试剂
呋喃唑酮代谢物:3-氨基-2-噁唑烷基酮(AOZ),呋喃它酮代谢物:5-吗啉甲基-3-氨基-2-恶唑烷基酮(AMOZ),呋喃西林代谢物:氨基脲(SEM),呋喃妥因代谢物:1-氨基-2-内酰脲(AHD),均购于德国Dr.Ehrenstorfer公司,纯度≥99%;硝基呋喃代谢物内标AOZ-D4,SEM-13C15N2,AMOZ-D5,AHD-13C3均购于德国Witega公司,纯度≥99%;乙腈:HPLC级,美国Merck公司;甲酸:LC-MS级,Fisher Scientific(中国)有限公司;2-硝基苯甲醛、盐酸、磷酸氢二钾、乙酸乙酯:AR级,国药集团化学试剂有限公司。
1.2 仪器与设备
Qtrap 6500三重四级杆质谱仪,美国AB SCIEX公司;Ultimate 3000液相色谱仪,美国Thermo Fisher公司;AcclaimTM RSLC 120 C18色谱柱(100 mm×2.1 mm,2.2 μm),美国Thermo Scientific公司;Elmasonic P超声仪,德国Elma公司;Allegra X-15R离心机,贝克曼库尔特有限公司;N-EVAP 116氮吹仪,上海安谱科学仪器有限公司;Milli-Q超纯水器,法国密理博公司;0.22 μm有机相微孔滤膜,天津市津腾实验设备有限公司;YALBOYS涡旋混合器,上海安谱科学仪器有限公司;ME204电子天平,梅特勒-托利多仪器有限公司。
1.3 方法
1.3.1 相关溶液的配制
(1)标准溶液的配制:分别准确称取AOZ、SEM、AMOZ、AHD标准品,用甲醇溶解,配制成100 μg/mL的单标标准储备液;移取适量单标标准储备液,用水稀释,配制成0.1 μg/mL的混合标准工作溶液,于-18 ℃冰箱中保存。
(2)内标溶液的配制:分别准确称取AOZ-D4,SEM-13C15N2,AMOZ-D5,AHD-13C3内标标准品,用甲醇溶解并定容,配制成10 μg/mL的单标标准储备液;移取适量单标内标标准储备液,用水稀释,配置成0.1 μg/mL的混合内标标准工作溶液,-18 ℃冰箱中保存。
(3)0.1%甲酸水溶液:准确吸取1 mL LC-MS级甲酸,用超纯水定容至1 L。
(4)0.1 mol/L 2-硝基苯甲醛衍生剂:称取1.5 g 2-硝基苯甲醛,用甲醇溶解并定容至100 mL。
(5)1.0 mol/L磷酸氢二钾溶液:称取87.1 g磷酸氢二钾,溶解于500 mL水中。
(6)0.2 mol/L盐酸溶液:量取浓盐酸17 mL,用水稀释至1000 mL。
1.3.2 样品制备与前处理
克氏原鳌虾样品,经去虾壳、虾线,取可食肉组织,绞碎混合均匀,于-20 ℃下避光保存,备用。
称取绞碎的样品2.0 g,置于50 mL离心管中,加入硝基呋喃代谢物混合内标工作溶液50 μL,涡旋混合50 s,再加入10 mL 0.2 mol/L盐酸溶液和100 μL 0.1 mol/L 2-硝基苯甲醛衍生剂,涡旋混匀50 s,将混匀后的离心管置于75 ℃恒温超声仪中超声提取反应80 min,取出离心管冷却至室温,加入2 mL 1.0 mol/L K2HPO4溶液,涡旋混匀50 s,加入10 mL乙酸乙酯,旋转混合20 min,4000 r/min离心5 min。上清液于40 ℃ N2吹干,残渣用2.0 mL流动相涡旋混合溶解,经0.22 μm滤膜过滤后上机测定。
1.3.3 液相色谱条件
色谱柱采用AcclaimTM RSLC 120 C18(100 mm×2.1 mm,2.2 μm),流动相A为乙腈,流动相B为0.1%甲酸水溶液,洗脱梯度程序为:0.0~2.0 min,B相保持90%,2.0~7.0 min,B相90%~5%,7.0~12.0 min,B相保持5%,12.1~15.0 min, B相90%。柱温40 ℃;进样量5 μL;流速0.4 mL/min。
1.3.4 质谱条件
采用电喷雾离子源;正离子模式;多反应监测扫描;喷雾电压5500 V;离子源温度550 ℃;雾化气流量(GS1):55 L/h;气帘气流速(CUR):35 L/h;辅助气流量(GS2):55 L/h;其他质谱参数见表1。

表1 4种目标物及4种内标物的优化质谱参数Table 1 Optimized parameters of MS/MS for 4 target and 4 internal standard compounds
1.4 数据处理
采用仪器配置的MultiQuant定量分析软件,对采集样品中目标化合物的定量离子对进行定量分析,采用化合物的定性离子对以及定性定量离子对丰度比进行定性分析。
2 结果与讨论
2.1 衍生方式选择
国家标准检验方法对硝基呋喃类药物的检测,前处理过程中的衍生步骤主要采用37 ℃恒温箱过夜16 h[18]或37 ℃恒温水浴避光振荡16 h[19],较为耗时,本研究比较超声波辅助衍生和恒温振荡衍生两种方式对硝基呋喃代谢物衍生效率的影响,设定温度60 ℃,衍生1 h,得到两种不同衍生方式下对应衍生产物的含量结果比较,见图2。试验结果表明,当采用标准溶液进行衍生反应时,两种衍生方式下衍生产物的含量相当,但采用阳性样品进行衍生比较试验时,超声波辅助衍生所得的衍生产物含量明显高于恒温振荡衍生方式下的产物含量,这可能是由于硝基呋喃代谢物在动物组织中主要以结合态形式存在,需要在酸性条件下将目标代谢物从组织蛋白中水解游离出来,才能与衍生试剂发生反应生成相应的衍生产物,超声波辅助衍生能加速不互溶两相之间的质量传递,从而加快硝基呋喃代谢物从组织结合蛋白中水解游离出来,并与2-硝基苯甲醛衍生试剂迅速反应生成相应的衍生产物,提高衍生效率。因此,选择超声波辅助衍生的方式进行样品前处理。

图2 超声波辅助衍生和恒温振荡衍生对硝基呋喃药物衍生效率的影响Fig.2 Effect of ultrasonic and constant-temperature oscillation on the derivatization efficiency of nitrofuran drugs
2.2 衍生温度的优化
在空白基质样品中添加适当浓度的混合标准工作溶液,按照1.3.2节样品制备方法进行前处理,并分别设置45、55、65、75、85、95 ℃进行衍生温度的比较和优化,结果见图3,从图3可以看出,当衍生温度为75 ℃时,四种硝基呋喃类目标化合物衍生效果较好,衍生温度为95 ℃时,AHD和AMOZ衍生效果变差,因此,选择75 ℃作为衍生最佳温度。

图3 温度对硝基呋喃药物衍生效率的影响Fig.3 Effect of temperature on the derivatization efficiency of nitrofuran drugs
2.3 衍生时间的优化
在选定衍生方法和衍生温度的前提下,通过空白基质样品加标的方式进行衍生时间的比较,分别设置25、40、50、60、70、80、135、160、210 min进行衍生时间比较优化,结果见图4,从图4中可以看出,当衍生时间为80 min时,4种硝基呋喃类化合物衍生效果最佳,因此选择衍生时间为80 min进行试验。

图4 时间对硝基呋喃药物衍生效率的影响Fig.4 Effect of time on the derivatization efficiency of nitrofuran drugs
2.4 色谱质谱条件的优化
采用1.3.2节样品前处理方法对4种硝基呋喃代谢物混合标准工作溶液和4种内标混合标准工作溶液分别进行衍生、提取、浓缩、复溶,对复溶后的衍生标准溶液,采用针泵以流动注射的方式进行各目标化合物质谱条件的优化,8种化合物均在正离子模式下具有较强的分子离子峰,因此在正离子模式下进行母离子全扫描,选定母离子,对母离子施加合适碰撞能量,使其产生子离子,并对子离子进行全扫描,选取丰度较强的两对子离子作为定性离子,并同时优化去簇电压(DP)、碰撞能量(CE),使灵敏度达到最佳,优化后的质谱条件见1.3.4节。4种硝基呋喃代谢衍生物及4种内标衍生物的MRM图如图5所示。
采用0.1%甲酸水溶液-乙腈作为流动相,进行色谱条件优化,改变流动相的洗脱梯度,比较各目标物在不同洗脱梯度条件下的分离度,发现在1.3.3节梯度条件下,8种目标化合物离子对的分离度、响应强度及色谱峰形均较好。另外,本研究中采用的色谱柱粒径为2.2 μm,分别设置流速0.2、0.3、0.4 mL/min,比较考察色谱图,并结合出峰时间和系统压力综合分析,发现流速为0.4 mL/min时峰形尖锐,且出峰时间在10 min内,因此选择0.4 mL/min流速进行试验。

图5 4种硝基呋喃代谢物及4种内标物质的定量离子色谱图Fig.5 Chromatograms of four nitrofuran metabolites and four internal standards in MRM mode

表2 4种硝基呋喃代谢物的线性范围、线性方程、相关系数、检出限Table 2 Linear ranges, linear equations, correlation coefficients (R2) and limits of detection (LODs) of the four nitrofuran metabolites
2.5 定性定量离子对选择

图6 样品中SEM 定性定量离子对Fig.6 Qualitative and quantitative ion pairs of SEM in samples
质谱分析中主要通过对样品离子质荷比的分析而实现对样品的定性和定量分析。表1质谱参数显示,4种硝基呋喃类代谢物均选择了两对子离子,一般情况下将灵敏度高的子离子做为定量离子,灵敏度低的子离子做为定性离子,本研究中除SEM外,其他化合物定性定量离子对选择均遵循此规律,详见表1,但在实际样品分析中,SEM的209.0/166.0离子对响应高于209.0/192.0离子对,但209.0/166.0离子对在低浓度下存在样品杂质干扰,见图6,从而导致定量不准确,而209.0/192.0离子对无干扰,因此选择209.0/192.0离子对作为SEM的定量离子对。
2.6 方法学验证
2.6.1 线性与检出限
配制混合标准系列工作溶液,加入混合内标溶液,采用1.3.2节样品前处理方法进行处理,得到衍生后的标准溶液,按1.3.3节色谱条件和1.3.4节质谱条件,进样分析,以目标化合物的质量浓度(ng/mL)为横轴(x),相应的定量离子的峰面积与内标峰面积的比值为纵轴(y),绘制标准曲线,得到线性回归方程,结果见表2,采用空白样品加标试验确定检出限,根据3倍信噪比进行计算,得到检出限结果见表2。
2.6.2 回收率和精密度

表3 4种硝基呋喃代谢物的加标回收率和精密度Table 3 Recoveries and RSDs of four nitrofuran metabolites spiked in samples
在基质中分别添加低、中、高3个质量浓度的混合标准溶液进行加标回收试验,每个加标水平重复6次,按照优化后的方法进行处理并检测,得到结果见表3,4种硝基呋喃代谢物的平均回收率为93.3%~104.9%,相对标准偏差<10%,表明该方法的准确性和重复性良好,可以满足克氏原鳌虾样品中硝基呋喃代谢物测定的分析要求。
2.7 实际样品测定以及与国标方法比较
采用所建立的方法,对采集的20批次克氏原鳌虾样品进行检测,并对检出阳性的样品采用国标方法进行结果比对,见表4,结果表明采用超声波辅助衍生的方法所测得的样品结果与国标方法所检测结果基本保持一致,超声波辅助衍生法能显著缩短样品处理时间,提高检测效率,在实际应用中具有一定的优势。

表4 阳性样品检测结果对比Table 4 Comparison of detection results of positive samples(n=2)
3 结论
本试验对克氏原鳌虾样品中的硝基呋喃类代谢物的检测方法进行研究,优化了衍生方式,以超声波辅助衍生代替国家标准中的恒温水浴振荡衍生,优化后的方法可加快样品中目标化合物的水解速率和衍生反应速率,该方法操作简单、重现性好、准确度高,适合于克氏原鳌虾样品中硝基呋喃类药物代谢物的快速测定。但由于目前的研究发现甲壳类水产品中存在非呋喃西林来源的氨基脲成分,导致食品安全监管部门在进行甲壳类水产品是否违法使用硝基呋喃药物的行为判定上存在争议,今后可开展研究,寻找硝基呋喃类药物代谢的特征性标志物,以实现对样品的科学判定。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
化工管理(2021年7期)2021-05-13
理化检验-化学分册(2020年5期)2020-06-15
中国食品(2020年9期)2020-05-26
农药科学与管理(2019年5期)2019-08-13
中国农资(2016年1期)2016-12-01
合成化学(2015年10期)2016-01-17
烟草科技(2015年8期)2015-12-20
化工进展(2015年3期)2015-11-11
质谱学报(2015年5期)2015-03-01
