
血管内皮糖萼在脓毒症急性肺损伤病理机制及诊断治疗中的作用
2021-05-21 03:30:26陈加弟龚迪易玉虎陈怿
解放军医学杂志 2021年4期
陈加弟,龚迪,易玉虎,陈怿
1暨南大学附属第一医院急诊科,广州 510632;2暨南大学附属第一医院眼科,广州 510632;3暨南大学附属东莞医院重症医学科,广东东莞 523900
脓毒症(sepsis)是指宿主对感染的反应失控而导致的危及生命的器官功能障碍[1]。脓毒症往往累及多个器官系统,且常因器官衰竭而导致患者死亡[2],其中肺组织受损尤为常见,发生率超过40%[3],在临床上常表现为急性肺损伤(acute lung injury,ALI)或急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)[4]。早在20世纪80年代,已有学者发现糖萼与肺泡渗出及间质水肿有着密切关系[5],但直到近10年,糖萼的具体生理结构功能及其在ALI病理机制中的作用才逐渐清晰。本文就血管内皮糖萼(vascular endothelial glycocalyx,VEG)在脓毒症ALI病理机制及诊断治疗中的作用进行综述。
1 VEG的结构与功能
VEG是位于血管内皮细胞管腔面细胞膜上的蛋白质-多糖复合物,层厚0.1~1.0 μm,由血管内皮细胞合成分泌,是构成血管内皮表面的重要结构,其主体成分为蛋白聚糖和糖蛋白。蛋白聚糖具有核心蛋白和带负电荷的糖胺聚糖(glycosaminoglycan,GAG)侧链结构。GAG的主要成分包括透明质酸(hyaluronic acid,HA)、硫酸乙酰肝素(heparan sulphate,HS)、硫酸软骨素、角蛋白和硫酸皮肤素等,其中HS的含量最高,占比达50%~90%。核心蛋白骨架主要包含多配体蛋白聚糖(syndecan)、磷脂酰肌醇蛋白聚糖(glypican)和基底膜蛋白聚糖等成分,是VEG发挥主要生理功能的重要结构基础。位于蛋白聚糖之下的糖蛋白主要由E-选择素、P-选择素、细胞间黏附分子(intercellular adhesion molecules,ICAM)和血管内皮细胞黏附分子(vascular cell adhesion molecule,VCAM)等具有影响血细胞黏附、游走和浸润,干预凝血、止血和纤溶功能的蛋白分子组成[6]。目前认为,VEG是微循环功能的重要调节器,对维持内皮细胞结构功能的稳定、抑制微血栓形成、调节微循环血流、调控血细胞与内皮细胞的作用、防止炎性细胞黏附、维护血管壁屏障功能的完整等均具有重要作用(图1)。糖萼生理结构完整性的破坏将直接造成相应组织器官出现以下改变:(1)广泛微循环血栓形成;(2)血管内皮细胞结构功能异常;(3)促进循环中炎性细胞滚动、黏附并游走至血管外的组织间隙;(4)血管壁完整性下降和血管通透性增加,加重组织器官水肿,导致细胞组织代谢障碍[7-8]。在各种病理因素的作用下,基质金属蛋白酶、乙酰肝素酶和唾液酸酶等的活性及浓度异常均可引发VEG损伤[9],该病理生理变化已被证实是诱发动脉粥样硬化、缺血/再灌注损伤和糖尿病并发症等病理损伤的始动因素[6]。
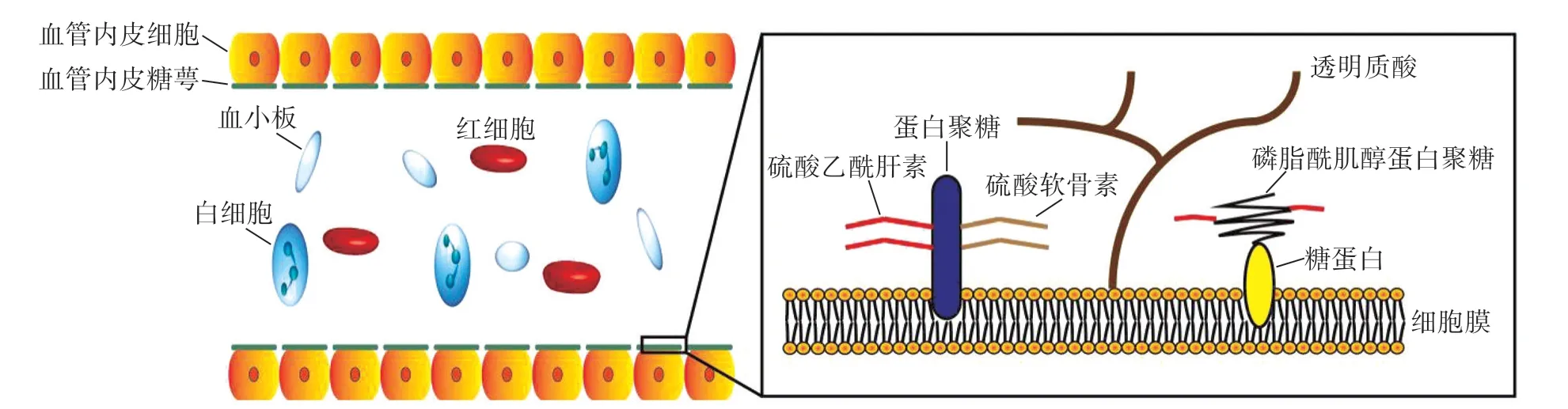
图1 VEG的主要结构及组成成分Fig.1 The main structure and components of VEG
2 肺VEG在脓毒症ALI病理机制中的作用
肺VEG在脓毒症ALI/ARDS发生发展过程中具有重要作用。在哺乳动物的组织器官中,与肺外血管(糖萼层厚0.6~0.8 μm)相比,肺泡毛细血管糖萼层更厚(可达1.5 μm以上),且HS更为富集[10-14]。这种糖萼在肺内分布的优势,与防止肺水肿和增强对外界抗原的耐受性等作用有关,因此在脓毒症发病过程中糖萼结构功能障碍也较其他器官更为严重,直接导致肺成为脓毒症首发打击或打击最为严重的器官。Inagawa等[15]通过腹腔注射脂多糖(lipopolysaccharide,LPS)建立小鼠脓毒症模型,在扫描电子显微镜和透射电子显微镜下观察小鼠肺VEG,发现注射LPS后48 h,VEG出现崩解破坏现象,表明肺VEG降解在脓毒症ALI/ARDS的发病机制中具有核心地位[12,15-16]。
2.1 脓毒症ALI/ARDS时肺VEG的损伤机制 在哺乳动物体内,生理状态下VEG处于合成与降解的动态平衡。Schmidt等[12]发现,脓毒症时炎性介质如白细胞介素-6(interleukin-6,IL-6)、肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、血管生成素-2(angiopoietin-2,ANGPT-2)等可使肺组织内乙酰肝素酶(heparanase,HPSE)的活性及浓度异常增高,进而加速糖萼结构的崩解[12,17]。HPSE是一种存在于哺乳动物体内的肝素酶Ⅲ类似物,由血管内皮细胞或中性粒细胞等体细胞分泌释放,可特异性分解HS,导致糖萼降解。此外,血管生成素(angiopoietins,ANGPTs)及其受体内皮细胞酪氨酸激酶型受体-2(endothelial tyrosine kinase receptor-2,Tie-2)也是调节糖萼合成与降解动态平衡的重要信号通路[7]。ANGPTs家族共包括4种亚型,起主要生理作用的ANGPT-1、ANGPT-2亚型大部分由血管内皮细胞和周细胞分泌产生,Tie-2是整个家族的共同受体,其中ANGPT-1/Tie-2的结合可促进糖萼形成与修复,维持血管内皮功能稳定,而ANGPT-2/Tie-2的结合可加速糖萼降解,造成血管内皮损伤。ANGPT-1与ANGPT-2之间存在数量竞争性抑制的关系,脓毒症ALI时,大量过表达的ANGPT-2可通过竞争阻断ANGPT-1与Tie-2的结合(图2),抑制Tie-2磷酸化[18-20]。此外,脓毒症时肺VEG的修复重建功能受到明显抑制,是促进ALI/ARDS进展的关键机制[21](图2)。Yang等[21]指出,成纤维细胞生长因子受体1(fibroblast growth factor receptor 1,FGFR1)/HS生物合成酶exostosin-1(EXT-1)信号通路是调控肺VEG修复的主要分子机制:糖萼重建源于糖萼崩解并释放出HS片段,HS可直接激活成纤维细胞生长因子2(fibroblast growth factor 2,FGF2)形成HS-FGF2复合物,激活FGFR1和EXT-1的表达,最终启动肺VEG修复程序。但在脓毒症小鼠模型中发现,FGFR1和EXT-1的表达下调,致使肺VEG重建出现明显的延迟和抑制[22]。值得注意的是,Yang等[21]和Oladipupo等[23]发现,由于FGFR信号的组织差异性,肺VEG修复先于其他系统内皮组织,甚至是在全身炎症反应仍较明显时。
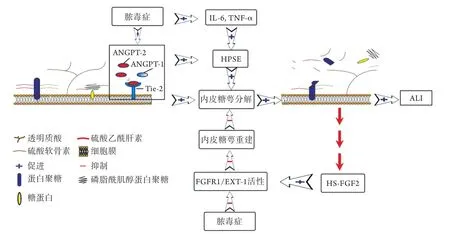
图2 脓毒症时肺VEG降解致ALI发生的模拟图Fig.2 Simulation diagram of ALI caused by VEG degradation in sepsis
2.2 肺VEG在肺损伤发生发展中的作用 目前认为,ALI/ARDS发病的细胞基础是肺血管内皮和肺泡上皮急性损伤,继而引发炎性细胞浸润和肺血管通透性增加[3,24]。VEG受损与炎症失控是ALI/ARDS的两个关键病理生理环节[25-26]。肺VEG降解不仅增加了肺毛细血管的通透性,形成以大量蛋白质渗出为主的肺水肿[27],还可显著提高肺毛细血管中性粒细胞的变形和黏附能力,使其更易游走至肺间质并释放炎性介质,在肺组织中形成“瀑布式”的炎症级联反应[28]。
首先,VEG是内皮细胞抵御损伤的防护屏障,可影响各种分子物质对内皮细胞屏障的穿透能力。VEG最浅层带负电荷,与血液中多种蛋白质产生静电斥力,阻碍白蛋白等大分子物质通过。因此,VEG损伤可直接导致静电斥力消失,加之内皮细胞间紧密连接随着VEG结构完整性被破坏而变得疏松,可造成血管通透性显著增加,血液中水分和血浆蛋白可外渗到肺间质和肺泡中,启动并加重肺水肿[27,29]。
其次,VEG在维持微血管生理功能中发挥着关键作用,可调节血管张力和凝血状态[28,30]。VEG与舒血管物质内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)的生成有关。Mochizuki等[31]、Kumagai等[32]和Yen等[33]先后证实,VEG参与内皮细胞机械应力(主要是血流引起的剪切应力)的传感与转导,进而促进eNOS的分泌释放,发挥调节血管张力和血流量、抑制血小板聚集和黏附等作用,所以肺VEG损伤可诱发肺毛细血管收缩和血流量减少。
第三,VEG损伤可激活并增强炎性细胞的活性,导致炎症反应失控。VEG裂解出的HS片段与LPS具有类似的作用,可通过激活依赖于MyD88的Toll样受体4(Toll-like receptor-4,TLR-4)将信号传导至下游的核因子-κB(NF-κB),导致IL-6、TNF-α等炎性介质的生成和释放增加。同时炎性介质浓度的升高又可促进HPSE的活化,从而导致VEG降解加速,产生更多的HS片段,形成闭环式的正反馈调节[34]。有研究发现,VEG的降解暴露出血管内皮细胞的表面黏附分子,致使中性粒细胞、单核-巨噬细胞等炎性细胞更易黏附于内皮细胞,并进一步跨内皮迁移至血管外的组织间隙中导致炎症播散[12,35]。生理状态下,HS和syndecan-1可与部分趋化因子相结合,而在ARDS状态下,VEG降解后释放出的趋化因子可直接导致中性粒细胞的募集和游走,进而形成“瀑布式”的炎症级联反应。
3 VEG在脓毒症ALI诊断治疗中的作用
VEG损伤是启动ALI/ARDS的关键扳机,在脓毒症ALI/ARDS诊断评估和指导治疗等方面具有重要价值。有研究指出,脓毒症ALI/ARDS患者外周血中HPSE含量和活性明显上调,并在外周血中检测到糖萼降解相关产物浓度增加[36]。通过检测VEG降解产物评估ALI/ARDS患者病情严重程度,并以VEG为作用靶点治疗肺损伤成为近年来糖萼相关研究的热点。
3.1 VEG作为脓毒症ALI/ARDS诊断标志物Syndecan-1在多个临床报告中已被用作反映糖萼损伤的标志物[15,37]。近期一项临床研究回顾性调查135例脓毒症合并ARDS患者发现,血浆syndecan-1水平与ARDS严重程度直接相关,且可预测发生肺外器官衰竭及死亡的风险[37]。HS是组成VEG的重要物质,有研究发现,在脓毒症合并ARDS患者体循环中HS水平呈明显升高的趋势,且其水平与病情严重程度密切相关。ARDS并发休克患者HS水平可升高至对照组的4倍,以90 d结局进行亚组分析,死亡组的HS水平为存活组的3倍[38]。作为哺乳动物体内唯一可水解VEG的酶,HPSE的浓度在脓毒症并发ALI[12]和肾衰竭[39]时可升高,在感染性休克时升高更为明显。值得注意的是,不同致病原感染所致的HPSE水平升高程度存在显著差异,其中革兰阴性菌感染患者的HPSE水平和活性最高[40]。
3.2 基于VEG的脓毒症ALI/ARDS治疗靶点 由于ANGPTs/Tie-2轴信号通路具有调控VEG生成和降解动态平衡的重要功能,有研究指出,作为ANGPTs家族中促VEG降解的ANGPT-2是脓毒症ARDS的一个新型生物标志物[18,41]。Han等[19]在小鼠脓毒症模型中运用单克隆免疫球蛋白ANGPT-2结合抗体(ANG2-binding and Tie2-activating antibody,ABTAA)阻断ANGPT-2/Tie-2的结合,使VEG破坏得到明显缓解,从而减轻了小鼠ALI并提高了其存活率。Margraf等[42]发现,输注6%羟乙基淀粉具有抑制HPSE和透明质酸酶释放的作用,可减少syndecan-1和HA的脱落,对VEG的完整性具有一定保护作用,可减轻脓毒症ARDS患者肺血管通透性的异常。
鞘氨醇-1-磷酸(sphingosine-1-phosphate,S1P)是一种鞘磷脂,通过作用于细胞表面G蛋白偶联受体S1P1发挥作用,对血管内皮具有一定的保护效应[43]。Zeng等[44]的体外研究发现,激活S1P并促进其与S1P1受体结合可有效抑制syndecan-1脱落,进而保护糖萼免受病理损伤。该团队的另一项研究证实,上述保护作用的完成需要一个关键步骤——S1P由红细胞释放并转运至血管内皮细胞,血浆中的白蛋白为该过程的主要运载工具[45]。因此,在脓毒症复苏期间使用白蛋白或新鲜血浆具有保护糖萼完整性及改善患者预后的作用[46-48]。
在脓毒症ALI发病机制中,肺血管内皮细胞分泌的HPSE扮演了启动并加速VEG降解的角色,因此抑制HPSE过度活化成为一种具有较好前景的脓毒症ALI/ARDS治疗方法[12]。Schmidt等[12]通过盲肠结扎和穿孔造模的小鼠脓毒症模型发现,单次注射肝素(在造模后24 h内)可抑制肺血管内皮通透性的异常增加,使用HPSE抑制剂可完全遏制脓毒症诱导的VEG破坏,而肝素可与内皮糖萼非共价结合,促使蛋白构象变化,巩固VEG结构并增强其功能,最终改善糖萼降解[12]。
4 总结与展望
近年来关于VEG的研究逐渐增多,VEG作为血管内皮细胞分泌合成的重要物质,参与维持内皮结构功能的生理活动。在病理状态下,VEG降解表明血管内皮发生损伤,与血管渗漏、间质水肿、炎症播散、氧化应激、血管收缩甚至弥散性血管内凝血等各种病理性损伤的发生直接相关。大量证据表明,VEG在脓毒症ALI中扮演着极为关键的角色,且以VEG作为靶点采取各种手段调控其异常可发挥显著的肺保护作用。现阶段大部分以VEG为切入点进行的脓毒症ALI研究基于细胞或动物实验,临床研究的数量、规模和质量参差不齐,影响了VEG作为脓毒症ALI诊断标志物和治疗靶点在临床的进一步推广应用,但未来其可能是导致脓毒症集束化救治策略如液体复苏、炎症抑制和血流动力学分析等方面发生根本性变革的关键因素。
猜你喜欢
中华养生保健(2020年4期)2020-11-16 01:31:40
中国中医急症(2019年10期)2019-05-21 07:20:46
现代检验医学杂志(2016年1期)2016-11-12 13:19:44
中华老年多器官疾病杂志(2016年9期)2016-04-28 08:52:44
山东医药(2015年16期)2016-01-12 00:40:08
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
医学研究杂志(2015年10期)2015-06-13 00:37:34
医学研究杂志(2015年11期)2015-06-10 06:44:03
中国老年学杂志(2013年15期)2013-09-13 01:48:56
中国中西医结合外科杂志(2013年3期)2013-03-11 20:04:59
