
以糖尿病酮症酸中毒为表现的遗传性血色病1例报告
2021-05-17 09:56胡滢月鲁利莎李贞茂李良平
临床肝胆病杂志 2021年5期
胡滢月,鲁利莎,李贞茂,李良平,雷 蕾
四川省医学科学院·四川省人民医院 消化内科,成都 610072
铁是人体内必须微量元素之一,正常成人中90%~95%的铁来源于体内衰老或破坏的红细胞中的血红蛋白被巨噬细胞吞噬所释放的铁;5%~10%来源于肠道吸收食物中的铁。血色病为我国临床罕见铁代谢缺陷病,现将本科收治的1例表现为糖尿病酮症酸中毒,经基因检测、肝组织活检、影像学等证实为遗传性血色病患者的临床资料报道如下。
1 病例资料
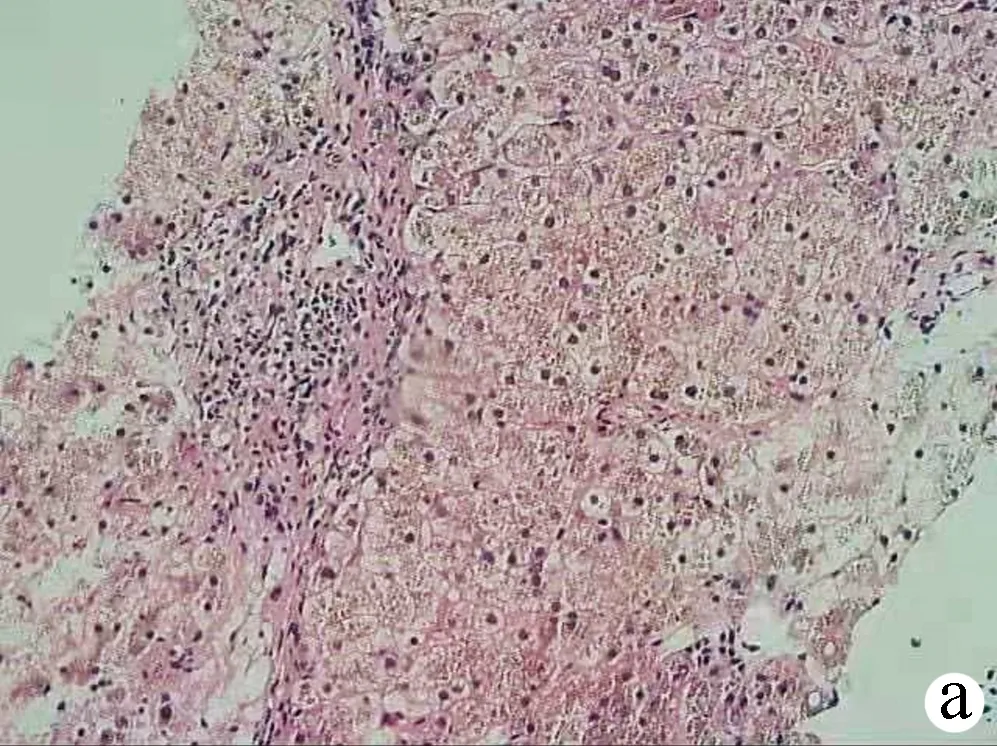
患者男性,31岁,因“肝功能异常7年,血糖升高2年,恶心、呕吐5 d”于2020年4月27日收入本科。既往有性功能减退1年,否认神经系统特殊病史,否认血液病、肝炎病史,否认长期口服铁剂史,否认有毒有害物质接触史,无输血史,无手术、外伤史,偶有饮酒史,未婚未育,家族无类似疾病病史,父母否认近亲婚育史。7年前体检肝功能:ALT 68 U/L,AST 94 U/L(未行诊治)。2年前体检空腹血糖为10.2 mmol/L,餐后2 h血糖为12.7 mmol/L;社区门诊多次空腹静脉血糖>7.0 mmol/L及餐后2 h静脉血血糖>12.0 mmol/L(具体数值不详),诊断为2型糖尿病,予以“门冬胰岛素30注射液(诺和锐30)14 U(早)及16 U(晚)皮下注射”控制血糖,治疗期间未定期监测血糖。5 d前因劳累后出现腹胀、恶心、呕吐,伴头晕乏力,社区门诊血糖仪查指尖随机血糖提示“Hi”,尿酮体(3+)、pH 7.21,诊断为“糖尿病酮症酸中毒”,给予补液支持、静脉滴注胰岛素降低血糖治疗,经治疗后腹胀症状持续存在,头晕乏力、恶心呕吐好转,完善腹部CT诊断“肝硬化、脾大”,遂于本科住院。入院查体,体温36.8 ℃,脉搏98次/min,呼吸20次/min,血压104/63 mm Hg,慢性病容,中等体型,神清语晰,全身皮肤巩膜无黄染,无肝掌及蜘蛛痣,颜面部及双下肢胫前皮肤见广泛褐色色素沉着(图1)。心肺(-)。全腹软,无压痛,无肌紧张及反跳痛,肝脾未触及,Murphy征阴性,移动性浊音阴性,肠鸣音3~4次/min。双下肢轻度凹陷水肿;病理征(-)。辅助检查,血常规: WBC 3.01×109/L,Hb 122 g/L,PLT 57×109/L;空腹血糖(静脉血)15.41 mol/L,糖化血红蛋白 7.09%;肝功能:AST 81 U/L、ALT 64 U/L、ALP、GGT、胆红素均正常。性激素:睾酮<0.45 nmol/L。尿常规:尿糖(++),酮体(-)。甲、乙、丙、戊型肝炎病毒抗体均为阴性,自身免疫性肝病相关抗体(6项+4项)均阴性。铜蓝蛋白、肿瘤标志物(AFP、CEA、CA125、CA19-9)、IgG及亚型、IgM均正常。贫血相关代谢产物:血清铁蛋白(SF) 5313.36 ng/ml,转铁蛋白饱和度(TS) 86.2%。心脏彩超示:三尖瓣轻度关闭不全。腹部增强CT示(图2): 肝左叶增大,肝叶比例失调,全肝密度弥漫性增高,肝内血管密度相对减低。诊断:肝硬化,门静脉高压、脾大,胰腺体积偏小,胆囊增大,盆腔少量积液。因患者有血糖升高表现及查体见皮肤色素沉着,辅查TS及SF明显升高,腹部CT提示“肝硬化”,故不排除原发性血色病。完善肝组织活检(图3):可见大量褐色铁颗粒沉着(4级),普鲁士蓝染色提示大量的小叶实质铁超负荷。进一步行血色病全基因检测(外送吴氏集团有限公司)。检验方法:sanger (先证者)全外显子组20检测,即对患者基因组DNA进行全外显子组捕获和测序,基于二代测序数据进行点变异及小片段插入缺失变异和大片段拷贝数变异分析,基因检测结果如下:(1)小片段插入缺失变异:突变基因HJV,突变位点chr1:145416617-145416618,突变信息:c.962-963delinsAA(P.Cys321*)纯合子;(2)点变异:突变基因HJV,突变位点chr1:145414799,突变信息:c.18 G>C(p.Gln6His)纯合子;(3)大片段拷贝数变异:染色体区带1q21.1,包含HJV至GRR89C之间的基因大片段缺失。
结合患者病史、临床及体征、肝活检结果及基因检测,排除血液病、长期大量输血、口服补铁药物等病因致继发性血色病,最终诊断:(1)遗传性血色病(2A型,3期);(2)肝硬化代偿期(Ⅰb期,Child A级),脾大伴脾功能亢进;(3)继发性糖尿病。予以序列性放血治疗400 ml/次,1次/周,至2020年9月2日患者放血治疗后感头晕乏力,查Hb为96 g/L,开始加用新型铁螯合剂“地拉罗司625 mg,2次/d”去铁治疗。于本院消化内科门诊定期随诊,动态监测Hb、SF、TS、肝功能等指标。经25次放血治疗联合口服地拉罗司2个多月后,2020年11月22日查SF由最初的5313.36 ng/ml降至1941.91 ng/ml。
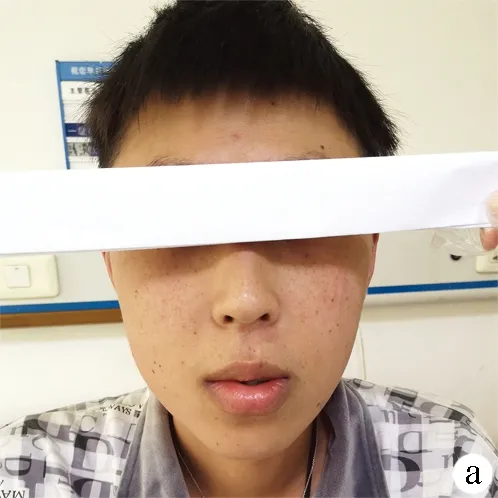


注:a,面部;b,双前臂及手背;c,双下肢胫前。


注:a,肝脏实质密度增高,呈“白肝症” ;b,肝内血管密度相对降低。
2 讨论
血色病根据病因分为原发性与继发性两大类[1]。原发性血色病又称为遗传性血色素沉着症(hereditary hemochromatosis,HH),由于基因突变使小肠铁吸收增加,铁在肝脏、皮肤、胰腺、心脏、生殖系统及关节内沉积,导致上述组织器官损伤、功能异常。继发性血色病(secondary hemochromatosis,SHC)多继发于溶血性贫血、无效造血的贫血、反复输血、慢性肝脏疾病。HH根据血色病基因(HFE、HJV/HAMP、TfR2、SLC40A1)突变情况分为4个类型:1型为HFE相关性血色病,以美国、加拿大及北欧国家的白种人多见;2、3、4型为非HFE相关血色病。不同基因型,铁沉积速度不同,临床表现不同。典型三联征包括:肝硬化、糖尿病、色素沉着。TS和SF联合检测可提高HH的检出率。TS是对于疑有铁过载患者的首选初筛方法,近年在大规模的人群筛查研究中发现不饱和铁结合能力在准确性上与TS有可比性,可做为筛查试验的替代[2]。SF敏感度高,正常则基本可排除铁过载[3],但因其易受多种因素(炎症、肿瘤、代谢综合征、慢性肝病)影响,故特异性差。影像学检查:CT表现为肝实质密度增高,呈“白肝症”;MRI呈“黑肝症”。但CT对于肝实质铁含量<150 μmol/g时密度变化不明显,且高密度铁易被脂肪肝的脂肪部分抵消出现假阴性,故MRI具有更高的敏感性和特异性,对血色病的早期诊断及治疗具有重要的临床意义[4]。肝组织活检:行HE及Masson三重染色进行组织学及肝纤维化分期;通过普鲁士蓝染色、肝铁浓度检测或肝铁指数进行铁的定量检测。最终确定诊断需靠血色病基因检测。





注: a,汇管区炎症,界面炎(HE染色,×100);b,肝细胞内可见较多色素沉着颗粒,肝细胞脂肪变性(HE染色,×200);c,CK19胆管上皮(+)(免疫组化,×100); d,CD68提示部分活化的Kupffer细胞(免疫组化,×100);e,网染提示肝板网状支架紊乱(×40);f,普鲁士蓝染色提示肝细胞内铁颗粒沉积(++++)(×100)。
2型血色病,亦称为幼年型血色病(juvenile hemochromatosis,JH),可分为A、B两个亚型,遗传方式均为常染色体隐性遗传。2型A亚型突变基因为HJV(铁调素调节蛋白基因),位于1号染色体长臂,编码血幼素,在加拿大、希腊、法国等的JH患者中2/3为G320V突变[5],而我国报道较多的突变形式为C321X[6]。2型B亚型突变基因为HAMP(肝脏抗菌蛋白)基因,位于19 号染色体长臂,编码铁调素。JH多见于近亲婚配家系,男女发病率大致相同,与典型HH相比,JH发病年龄早,临床症状多在30岁前出现[7],病情进展快,机体铁负荷和实质脏器功能损害程度重,更易表现为心肌病变、糖耐量降低和性腺功能降低,而非严重的肝脏疾病。不同类型血色病首选治疗方式为静脉放血,400~500 ml/次,1次/周,使SF浓度维持在50~100 ng/ml[8-9]。对于合并严重贫血低蛋白血症患者,亦可选择口服新型铁螯合剂,如地拉罗司,但需警惕剂量依赖性毒副作用,如胃肠道症状、皮疹、转氨酶增高、肌酐增高、听力减退等。
本例患者不明原因肝功能异常及血糖增高已几年,均未进一步查因,直至出现糖尿病酮症酸中毒后方逐步明确诊断。HH作为少见病,早发现早治疗有利于提高患者生存质量及延长生命,但该疾病确诊需依赖肝组织活检及基因检测,在多数基层医院无法开展上述检查,故基层临床医生需加强对血清TS及SF异常升高的意义及血色病影像学上的典型表现的认识,对上述人群及血色病患者的一级家属及早行基因筛查。本例患者的遗憾之处在于该患者父母拒绝行血色病相关基因筛查,故无法了解其族谱情况。我国HH基因突变位点以非1型HH为主[10],故仅行C282Y及H63D位点检测容易漏诊,建议行血色病全基因检测。本例患者因序列性放血治疗血清铁蛋白下降不满意,加用地拉罗司口服治疗,尚待进一步观察其远期疗效,积累治疗经验。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:胡滢月、鲁利莎参与病史收集,撰写及修改论文;李贞茂、李良平、雷蕾等负责拟定写作思路,指导撰写文章并最后定稿。
猜你喜欢
保健医苑(2022年6期)2022-07-08
家庭科学·新健康(2022年3期)2022-05-10
保健医苑(2022年4期)2022-05-05
疯狂英语·新读写(2021年10期)2021-12-07
党员生活(2020年6期)2020-07-04
奥秘(2019年8期)2019-08-28
环境(2018年10期)2018-12-15
Coco薇(2017年7期)2017-07-21
小猕猴智力画刊(2016年6期)2016-05-14
科学启蒙(2009年10期)2009-11-16
