
使用离散增量结合支持向量机方法预测蛋白酶的类型
2021-05-13 08:36王婷
农业与技术 2021年8期
王 婷
(长治职业技术学院,山西 长治 046000)
本文使用chou和shen相同的数据库[3],从蛋白酶的一级序列出发,以氨基酸组分,氨基酸二肽组分和亲疏水三肽组分为参数,采用离散增量结合支持向量机(ID-SVM)的方法对蛋白酶的类型进行预测,获得了较好的预测效果。
1 材料和方法
1.1 数据来源
本文选取了chou和shen创建的蛋白酶数据库[4](http://merops.sanger.ac.uk/(version 8.1,released on 05-May-2008))。此数据库的特点:序列同源性小于25%;序列长度不小于50个氨基酸;去除所有含有未知残基的序列。基于以上标准,最终得到3051个蛋白酶序列,可以分为6个类型:258个丝氨酸蛋白酶(aspartic)序列;589个苏氨酸蛋白酶(cysteine)序列;39个半胱氨酸蛋白酶(glutamic)序列;1040个天冬氨酸蛋白酶(metallo)序列;1063个金属蛋白酶(serine)序列;62个谷氨酸蛋白酶(threonine)序列。本文以这6类蛋白酶的一级结构为研究对象。
1.2 特征参数
1.2.1 氨基酸n肽组分
肽是构成蛋白质的结构片段,也是蛋白质发挥作用的活性基础部分。当n=1时,氨基酸n肽组分就退化为氨基酸组分,即20种氨基酸出现的频数,其表达相对简单,但丢失了各氨基酸间的关联信息。当n=2时定义为二肽组分,即400种氨基酸二联体出现的频数,此时加入了氨基酸间的排列次序和关联信息[5]。以此类推,n个氨基酸缩合成的n联体就称为n肽组分。
1.2.2 氨基酸序列的亲疏水性分布
蛋白质是由20种不同的氨基酸组成的生物大分子,蛋白质分子中的氨基酸残基靠酰胺键连接,形成含多达几百个氨基酸残基的多肽链,不同类型的氨基酸所包含的侧链结构和性质也各不相同,因此类型不同的氨基酸具有不同的物理化学性质。大量实验证实,蛋白质序列中氨基酸的物理化学性质也是影响蛋白质结构和功能的重要因素,尤其是分子结构中的亲疏水性分布的特征。
本文选取了氨基酸的亲疏水性分布作为描述氨基酸序列的一个信息参数。根据Li F M等、Chen Y L等、Pánek J等的方法对20种氨基酸进行分类,按照单个氨基酸亲疏水性分布的不同,将20种氨基酸分为以下6类:强亲水性类、强疏水性类、弱亲水或弱疏水性类、脯氨酸、甘氨酸、半胱氨酸。具体分类如表1所示。
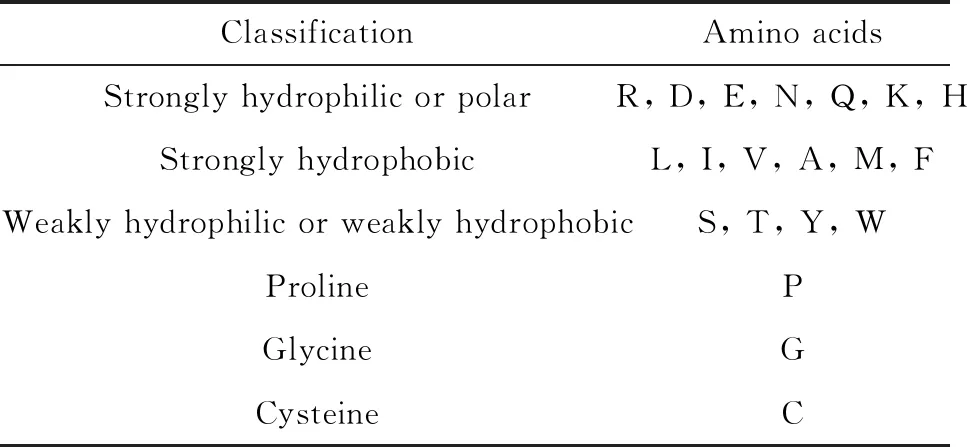
表1 氨基酸的亲疏水特征分类
1.3 预测方法
1.3.1 离散增量算法(increment of diversity, ID)
云计算是一种利用大规模低成本运算单元通过IP网络相连而组成的运算系统,用以提供各种计算和存储服务。由于具有高性能、低成本、可平滑扩展等优势,云计算为IT技术发展提供了新的技术手段和业务模式,不仅是企业降低建设和维护成本的重要手段,更为企业技术、业务和管理创新带来了新的契机。
离散增量算法是一种公认的较好模式识别分类器。此算法近年来已成功应用于蛋白质亚细胞定位[9]、蛋白质折叠子的结构类型[10]及蛋白质超二级结构的识别[11]等工作。
对离散量给出如下定义,对于由s维信息符号构成的状态空间X,这里用mi表示第i个状态出现的个数,其离散源X{m1,m2,…,ms}中的离散量:
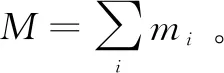
2个同为s维状态空间的离散源X:{n1,n2,…,ns}和Y:{m1,m2,…,ms},二者离散量:
混合离散源X+Y(n1+m1,n2+m2,…,ns+ms)的离散量:
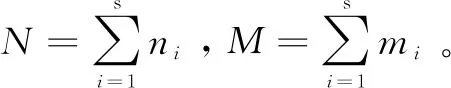
离散源X与Y的离散增量定义:
ID(X,Y)=D(X+Y)-D(X)-D(Y)
ID(X,Y)值用于比较2个离散源的相似性程度。ID(X,Y)值越小,表明2个离散源越相似;反之ID(X,Y)值越大,表明2个离散源的差异越大。
1.3.2 支持向量机算法(support vector machine, SVM)
支持向量机算法是由Vapnik等人于1995年提出的,具有相对优良的性能指标。此方法是建立在统计学理论基础上的机器学习方法。通过学习算法,SVM可以自动找出那些对分类有较好区分能力的支持向量,由此构造出的分类器可以最大化类与类的间隔,因此有较好的适应能力和较高的分辨率。该方法只需由各类域的边界样本的类别来决定最后的分类结果[12]。目前支持向量机算法在亚细胞定位[13,14]、蛋白质结构[15]和蛋白质相互作用[16]等方面都有广泛应用,其计算结果已经远远超过了各种传统的学习算法。支持向量机算法已经被很多学者编译为程序加以应用,这里使用的是由Chang和Lin联合开发的libsvm程序包,可以从网站(http://www.csie.ntu. edu.tw/~cjlin/)免费下载获得。
1.3.3 离散增量结合支持向量机算法(ID-SVM)
如果直接将从一级蛋白质序列中提取到的特征参数,输入支持向量机算法中,那么输入向量的维数就会非常大,势必造成维数灾难,使得支持向量机算法的优越性大打折扣。为了解决这一问题,提出了离散增量结合支持向量机算法(ID-SVM):将蛋白质序列中提取到的特征参数输入离散增量算法,将得到的离散增量值作为特征参数输入支持向量机中进行分类预测。这样做可以很好地降低支持向量机的输入向量维数,避免SVM过训练。
2 结果与讨论
本文选取了20个氨基酸组分,400个氨基酸二肽组分和216个亲疏水三肽组分作为离散源参数。每组参数得到的离散增量值均可以构成一个6维向量;再将这3组离散增量值组合起来,构成一个3×6=18维的特征向量输入支持向量机中进行分类预测。本文采用jacknife检验方法对蛋白酶的类型进行预测,其结果如表2所示。
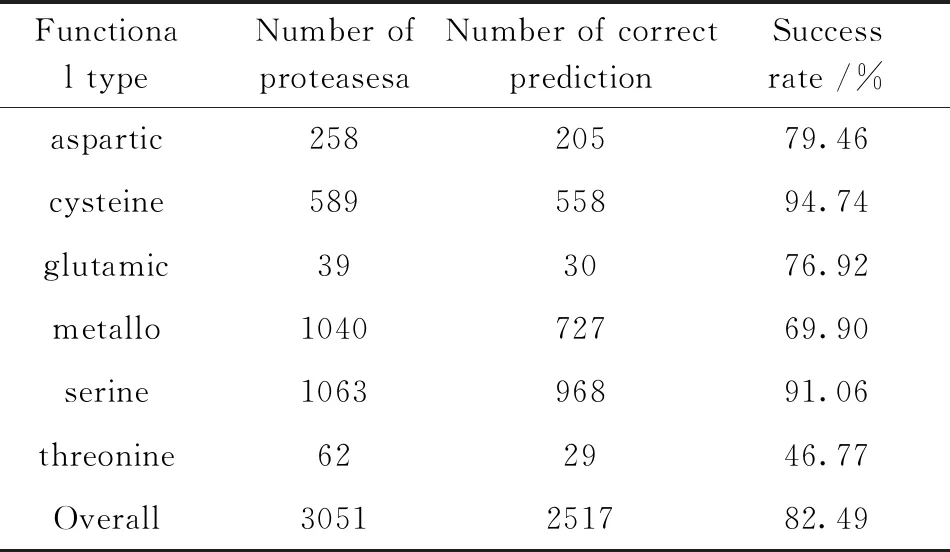
表2 基于ID-SVM方法在Jackknife检验下6种类型蛋白酶的预测结果
由表2可以看出,Jacknife检验方法的总体预测成功率为82.49%。相对而言,对第二类苏氨酸蛋白酶(cysteine)和第五类金属蛋白酶(serine)的预测效果更好一些,分别达到了94.74%和91.06%,而对第六类谷氨酸蛋白酶(threonine)的预测效果稍差。表明蛋白质序列数目越多的类,预测成功率可能会更高。如果能够进一步提取出更多更好的序列特征信息,预测结果肯定会提升。
3 结语
本文使用离散增量结合支持向量机方法预测蛋白酶的类型,能够获得较好的预测结果,有以下2个原因:离散增量算法可以有效降低参数向量的维数,简化计算过程;支持向量机算法能够很好的融合各种有益序列信息,并通过调节计算参数得到最佳的预测结果。
猜你喜欢
当代陕西(2022年6期)2022-04-19
煤气与热力(2021年12期)2022-01-19
成都大学学报(自然科学版)(2021年1期)2021-05-22
文苑(2018年22期)2018-11-19
中成药(2018年8期)2018-08-29
中成药(2018年2期)2018-05-09
电信科学(2016年9期)2016-06-15
西南军医(2016年6期)2016-01-23
电子设计工程(2015年16期)2015-02-27
食品工业科技(2014年7期)2014-03-11
