
基于集成学习的肿瘤药物敏感性预测研究
2021-05-07 08:42黄鹏杰林勇张梦欢吕琳刘振浩裴潇倜许林锋谢鹭
中国医学物理学杂志 2021年4期
黄鹏杰,林勇,张梦欢,吕琳,刘振浩,裴潇倜,许林锋,,谢鹭
1.上海理工大学医疗器械与食品学院,上海200093;2.上海生物信息技术研究中心,上海201203;3.中国科学院分子细胞科学卓越创新中心,上海200031
前言
肿瘤药物敏感性预测研究是精准医学的重要分支。当前,肿瘤精准医学不断发展,基于生物标志物和临床病理特点的抗肿瘤靶向治疗方法得到普遍应用[1]。在合适的时间对不同癌症患者采取个性化预防、治疗和用药指导,能使某些类型肿瘤的客观缓解率和5年生存率均有较大程度提高,同时对癌症病人的副作用比传统方法小,这是精准医学在肿瘤领域应用的成功典范[2]。
近年来,人工智能因为底层技术突破和海量数据驱动而不断发展,而生物信息领域随着第二代测序技术的发展产生大量的组学和药物反应数据,将两者结合能极大辅助肿瘤精准医学的发展。基因组学研究不断深入,大量研究表明肿瘤的产生、分化以及药物治疗和基因密切相关[3],通过基因表达谱来预测肿瘤药物的敏感性应答成为当下热点。Geeleher等[4]通过乳腺癌的细胞系和药物数据,建立了以岭回归(Ridge regression)算法为基础的基因表达和药物敏感性的回归模型;Menden等[5]利用CGP数据集,通过神经网络算法来建立药物应答预测模型;基于随机森林算法,Riddick 等[6]利用NCI-60 数据训练回归模型来预测药物敏感性;Gupta 等[7]研究了基于支持向量机和多种基因组类型的回归方法。
这些方法不仅促进了癌症药物基因组学的发展,也为预测药物敏感性提供了有效途径,但当前研究缺少对大批量药物和细胞系基因数据的有效处理,同时模型所采用算法不多且精准度还不够高。本文工作较上述研究具有以下特点:(1)药物种类多,且对细胞系和基因特征有效筛选用来建模;(2)采用集成学习算法,大部分药物预测准确率高,相比单模型有显著提升;(3)对特定药物进行功能分析,从生物学角度提供模型的可解释性。
本文基于GDSC2 数据库中809 个肿瘤细胞系样本的RNA-seq 数据和183 种药物与相关细胞系反应的IC50 值,经过样本筛选和基因特征降维后,结合boosting集成和4种机器学习方法来构建药物敏感性分类预测模型。实验结果表明,AdaBoost+SVM 集成方法相比单模型和其他集成模型,在整体药物集上的ROC 曲线下面积(AUC)、精准度(Precision)、准确率(Accuracy)、查全率(Recall)、综合评价指标(F1)等指标中具有更好效果。同时本文以药物Nutlin-3A(-)为例进行特异性分析,通过特征排序筛选和富集分析,从生物学和统计学验证了该药物的作用靶向通路P53。综上,本文的模型能对肿瘤病人的临床精准用药起到指导和建议作用。
1 基于集成学习的肿瘤药物敏感性预测方法
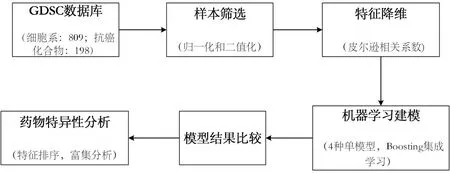
图1 分类预测方法流程图Fig.1 Flowchart of classification prediction method
1.1 数据集和预处理
本研究数据集来源于肿瘤药敏多组学数据库(Genomics of Drug Sensitivity in Cancer,GDSC),该数据库有多于1 000 种人类癌症细胞系的基因组特征和它们对不同抗癌药物的敏感性实验数据,是癌症细胞药物敏感性和药物反应分子标志物信息的最大公共资源。GDSC 目前由GDSC1 和GDSC2 两个数据库组成,后者主要收录2015年至今的测序和试验结果,它使用代谢测定法(CellTitreGlo)来确定细胞活力,是基于改进的技术、设备和程序等得到的最新数据[8]。
本文的数据集为GDSC2,包含有809 个细胞系,198 个抗癌化合物,以及135 242 个药物和细胞系反应数据。其中药物反应指标为IC50 值,即半抑制浓度,表示凋亡细胞与全部细胞数之比等于50%时所对应的药物浓度,反应细胞对药物的耐受程度。IC50 值越低,则说明细胞对药物越敏感。此外,为了精确匹配相关细胞系的基因表达值,笔者从ArrayExpress网站获得编号为E-MTAB-3610的数据,它包含GDSC 中所有细胞系的原始基因芯片[9]。接着,使用R 包Affy 读取CEL 格式文件并提取为基因表达矩阵,同时利用该包中的Robust Multi-array Average(RMA)函数对原始数据进行标准化,以达到背景校正、分位数标准化、对探测强度进行log 转换的目的。RMA标准化后,得到1 018个细胞系对应的17 737 个基因表达值,其中去除基因名重复和空缺的数据后,还剩17 418个基因做后续分析。
1.2 细胞系样本筛选和特征基因降维
对单个药物处理来说,本文先对细胞系实验样本的IC50值进行归一化处理:先计算均值,每个IC50值减去均值后再除以方差。然后对归一化后的数据由小到大进行排序,前后20%占比的样本分别视为有或无药物应答反应,同时去除待验证的中间样本,将预测模型从回归变为二分类问题。接着将每种药物筛选后的数据集进行8/2 分,分别用来训练和验证。此外,不同药物实验对应的样本数从48 到809不等,经过以上步骤处理后有15 种药物的样本数小于20,样本过少不利于建模和十折交叉验证,因此这15种不在本次研究范围。
接着,通过R语言匹配细胞系样本的基因表达值作为待输入特征,但选取的特征基因数过多,会提高计算复杂度、降低预测准确性,因此对特征降维极其重要。本文将待训练样本归一化后的IC50值和单个基因的表达值进行皮尔逊相关性检验,通过R 中的Cor 函数选出相关系数绝对值大于0.3,P值小于0.05的基因。最后,对不同药物来说,特征基因数从17 418降维到几十至几千不等。
1.3 基于集成学习的药物敏感性预测建模
1.3.1 单模型建模本研究使用的单模型是4 种经典的机器学习模型,分别为支持向量机(Support Vector Machine,SVM)、随机森林(Random Forest,RF),极端随机树(Extremely Randomized Trees,ET)和基于Bagging 的决策树(Decision Tree Based on Bagging,BT)。其中,本研究集成的主要基分类器为SVM。
SVM是基于结构风险最小化和VC维理论的监督式学习方法,通过构造分隔面将数据进行分离,在解决非线性、高维模式识别和大样本分类中表现出独特优势[10]。建模中,给定的肿瘤药物预测的样本集{(xi,yi),i=1,2,…,n},xi表示预测因子基因特征向量(L维),yi表示某细胞系对某药物是否有应答。对于这种高维非线性预测问题,SVM通过将原始空间映射到高维特征空间来建立分类预测模型。分为以下几步:(1)建立初始超平面:w·φ(xi)+b=0,其中,w为权值矢量,b为阈值,φ为目标函数。(2)寻找最优超平面,其可以归纳为求解如下优化问题:s.t.(w·φ(xi)+b)-1+ξi≥0,ξi≥0,其中C为惩罚参数,ξi为非负松弛因子。(3)接着求解得到最优分类决策函数:,其中,为最优拉格朗日乘子,b*为分类阈值且为核函数,sgn(*)表示返回整型变量的函数,xj表示不同于xi的样本点。
RF、ET、BT 都可看做Bagging 集成的变种,本文将其视为Boosting 集成的基分类器。RF 将bootstrap重抽样方法和决策树算法相结合,能对重要特征进行排序,是性能良好的用于分类或回归的有监督算法[11]。ET 与RF 相似,都由许多决策树构成,但它是完全随机得到分叉值进而对回归树分叉,不同于RF在一个随机子集内得到最佳分叉属性[12]。此外,该方法每棵回归树都使用全部训练样本。BT是完全用Bagging分类器集成决策树的方法。
[教学片段4]执教老师在讲不同国家赠送礼物的文化时,提到朋友结婚送礼的问题,让学生给她建议。她设计了三类情况,中国人的婚礼、英国人的婚礼和中国女生与英国男生的婚礼,通过追问帮助学生认识赠送礼要随当地的习俗,可以帮助学生理清关键信息的内涵。提问如下:
1.3.2 Boosting 集成学习建模AdaBoost 是最常见的一种Boosting 算法,能有效应用在两类、多类单标签等分类问题中,具有较强自适应能力、速度快和算法复杂度低等优点[13]。其主要思想是给每个训练样本赋予一个初始权值,再根据每次训练的样本分类是否正确以及总体准确率,来自动调整权值。接着将调整后的新数据集传给下个分类器,最后对每次训练的基分类器进行加权组合得到最终决策分类器。
本研究将该方法用在肿瘤细胞系对药物敏感性的分类中。设输入的训练样本为:{(x1,y1),(x2,y2),…,(xi,yi),…,(xn,yn)},其中xi中表示输入的带有基因表达信息的细胞系样本,yi∈{0,1}分别表示正样本和负样本,其中正样本数为l,负样本数m,n=l+m。具体步骤如下:
(1)初始化每个细胞系样本的权重wi,i∈D(i),在每个弱分类器t中(t=1,…,T,T为总个数),其权重可以归一化为一个概率分布:

(2)对每个特征训练一个弱分类器hj。由于本研究为二值分类,可通过遍历得到阈值,该阈值使错分的样本最小且错误率最低。接着计算包含所有特征的弱分类器的加权错误率:
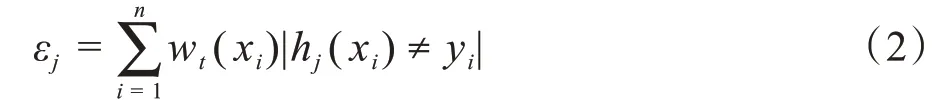
(3)选取错误率εt最小的弱分类器ht,并按照此最佳ht调整权重:

其中,εi=0 表示被正确地分类;εi=1 表示被错误地分类。

(4)加权得到最终的决策强分类器:
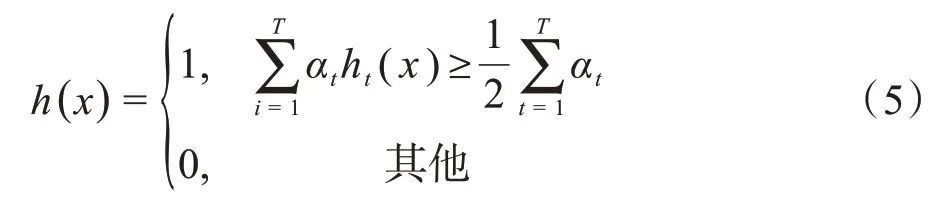

其中αt的计算公式为:经过以上一系列的迭代和集成处理后,我们可以得到药物敏感性预测的最终强分类器,来判定肿瘤细胞系能否对该药物产生应答反应。AdaBoost 默认的基分类器为决策树,本研究分别将SVM、RF、ET、BT 作为基分类器进行同质集成。基分类器个数对预测精度会有影响,本文分别设置为3、10、20、50进行实验,发现个数为10时达到理想效果,即使增加到50 准确率没有明显提高,反而会加大内存消耗和训练成本。因此,本文基分类器数量定为10。
1.3.3 十折交叉验证交叉验证(Cross Validation),也称循环估计,是一种统计学上将样本切割成较小子集的使用方法,常用于评估统计分析、机器学习算法对独立于训练数据的数据集的泛化能力[14]。
十折交叉验证(10-fold cross validation),将数据集分成10 份,轮流将其中9 份做训练,1 份做验证,10次结果的均值作为对算法精度的估计。本研究建模过程中均使用了十折交叉验证求均值的方法,其优势在于同时重复运用随机产生的子样本进行训练和验证,以求药物敏感性预测结果更精准。
2 实验验证和结果分析
2.1 实验评估指标
为了评估模型好坏,对比4 种分类模型的预测精度和泛化能力。本研究采用以下常用评价指标:AUC,Precision,Recall,F1值,Accuracy。在二分类中,“正例(positive)”和“负例(negative)”指分类标签,而“真(true)”和“假(false)”指模型预测结果是否与测试标签相对应。可知,TP、TN 表示分类结果正确;FP、FN 表示分类结果错误。各项指标的定义如下:

其中,TP 为真阳性,表示实验细胞系样本有药物应答,预测模型中也有。FP 为假阳性,表示实验细胞系样本无药物应答,但预测模型中有。FN为假阴性,表示实验细胞系样本有药物应答,但预测模型中没有。ROC 曲线在坐标系中越偏向左上角,即AUC 越大,则表示分类器的性能越好。
2.2 实验结果与分析
2.2.1 药物模型整体比较针对183 种药物的建模结果,本文详细比较了Boosting 集成方法和4 种单模型的各项评估指标。由图2 所有药物的AUC 分布情况可知,AdaBoost+SVM 集成模型有108 种药物集的AUC>0.9,AUC 在>0.95、0.9~0.95、0.85~0.9、0.8~0.85、0.75~0.8、<0.75 等区间的药物数占比依次为7.1%、51.9%、24.6%、11.4%、3.8%和1.1%,总体效果明显优于SVM、RF、ET、BT 和它们的集成模型。此外,其他3 种集成模型与各自的基学习器相比在大部分药物的准确率上也有所提升。
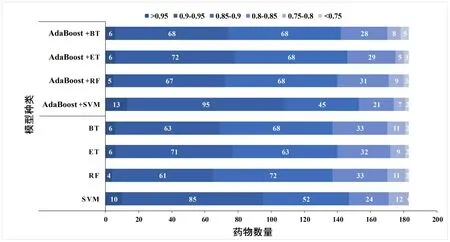
图2 183种药物在单模型和boosting集成模型的AUC分布情况Fig.2 AUC distribution of 183 drugs in the models based on a learner only and Boosting ensemble models
得到整体分析结果后,笔者选取在所有模型中都取得较高准确率的药物做进一步分析,分别为Venetoclax、OSI-027、Nutlin-3a(-)、AZD5991、Vorinostat、Oxaliplatin、Dasatinib、 Niraparib、 Paclitaxel、 Daporinad、Camptothecin、Cisplatin、MIRA-1。图3是这13种药物在集成模型和SVM中的AUC对比图,可知,AdaBoost+SVM模型的预测效果最优,且AUC都大于0.95。值得注意的是,对单个药物来说,取得最优效果的不一定都是AdaBoost+SVM,其他集成方法在个别药物数据集中也可能成为最优建模方法。
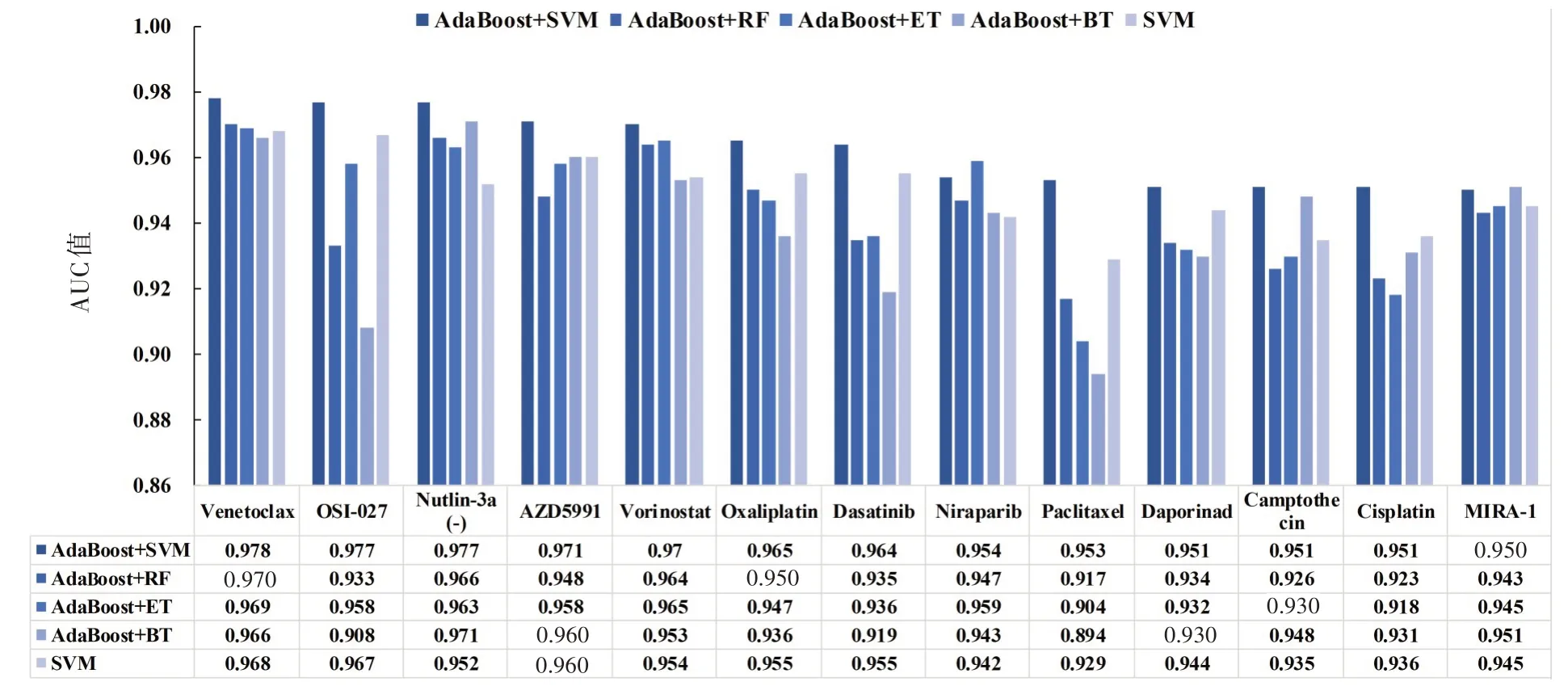
图3 高准确率药物预测模型比较Fig.3 Comparison of high-accuracy drug prediction models
为了更详细展示每种药物在不同方法建模中的效果,笔者以药物Nutlin-3a(-)为例进行分析。表1是该药物在Boosting 集成模型和4 种单模型中的AUC、Accuracy、Precision、Recall以及F1值的具体数值和标准差。图4为不同的模型进行十折交叉训练的ROC曲线,可以直观发现该药物在AdaBoost+SVM中效果最好,整体准确率相比单模型高出约2.5%,相比其他集成模型高出约1.5%,且各项指标的标准差相对最小。
表1 药物Nutlin-3a(-)在所有模型中的结果比较(±s)Tab.1 Comparison of prediction results of Nutlin-3a(-)in all models(Mean±SD)
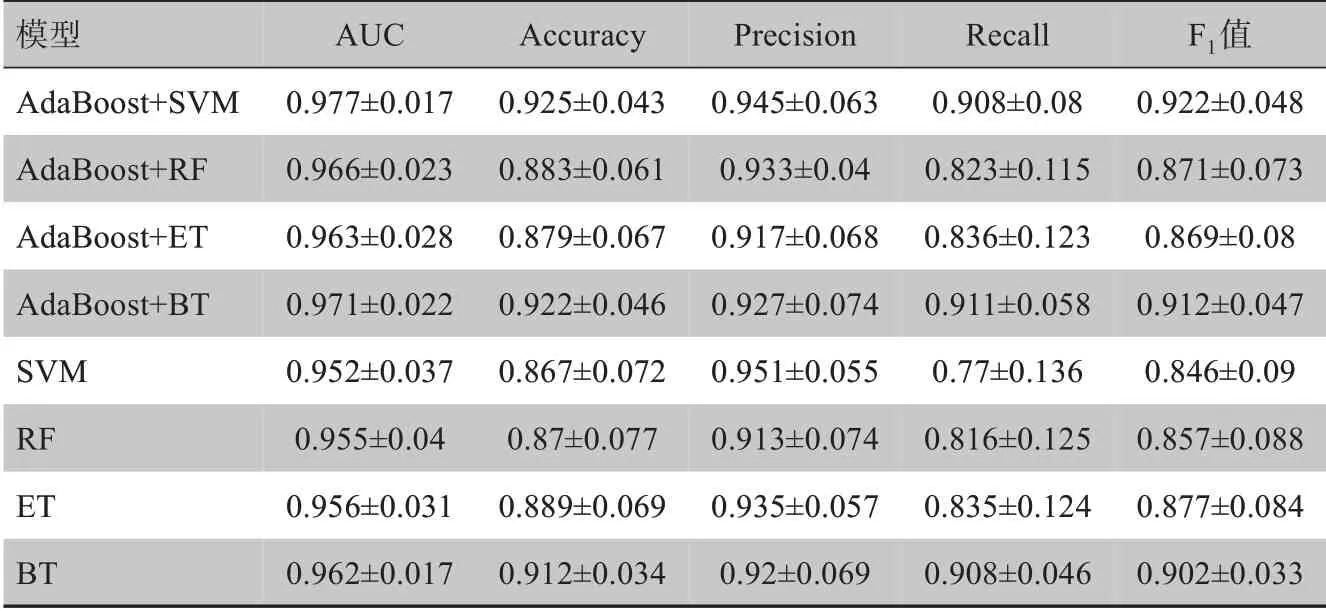
表1 药物Nutlin-3a(-)在所有模型中的结果比较(±s)Tab.1 Comparison of prediction results of Nutlin-3a(-)in all models(Mean±SD)
模型AdaBoost+SVM AdaBoost+RF AdaBoost+ET AdaBoost+BT SVM RF ET BT AUC 0.977±0.017 0.966±0.023 0.963±0.028 0.971±0.022 0.952±0.037 0.955±0.04 0.956±0.031 0.962±0.017 Accuracy 0.925±0.043 0.883±0.061 0.879±0.067 0.922±0.046 0.867±0.072 0.87±0.077 0.889±0.069 0.912±0.034 Precision 0.945±0.063 0.933±0.04 0.917±0.068 0.927±0.074 0.951±0.055 0.913±0.074 0.935±0.057 0.92±0.069 Recall 0.908±0.08 0.823±0.115 0.836±0.123 0.911±0.058 0.77±0.136 0.816±0.125 0.835±0.124 0.908±0.046 F1值0.922±0.048 0.871±0.073 0.869±0.08 0.912±0.047 0.846±0.09 0.857±0.088 0.877±0.084 0.902±0.033
2.2.2 药物特异性分析本文在所建模型基础上以Nutlin-3a(-)为例进行了药物特异性分析。从GDSC数据库可知该抑制剂已验证的药物靶点为MDM2,作用通路为P53。如图5所示,本文通过随机森林模型选取该药数据集的重要特征基因进行富集分析,从生物学和统计学两方面验证了该药物的P53 靶向通路[15]。该生物功能分析为模型提供了可解释性和生物理论支撑。
3 总结与展望
本文针对GDSC2 中的183 种抗癌药物和809 个细胞系数据,经过有效的样本筛选和基因特征降维后,首次使用AdaBoost+SVM集成方法建模来预测不同细胞系对药物的敏感性。此外,通过比较AUC、Accuracy、Precision、Recall 和F1值后,发现该模型相比4 种单模型和其他集成方法在整体药物数据集中效果最优。与现有研究的模型相比,不仅具有较高精确度,而且通过药物特异性分析为模型提供了生物可解释性。以上研究证实了将集成学习方法应用在肿瘤药物敏感性预测的可行性,能在临床用药的筛选中发挥作用。实际应用中,可以先对病人的肿瘤组织进行基因测序,再使用模型去预测特定抗癌抑制剂对该病人是否有效,以达到提高用药疗效和减轻副作用的目的[16]。
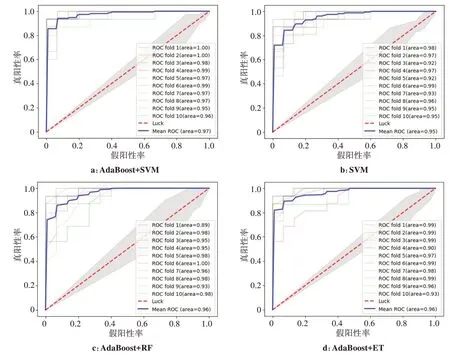
图4 Nutlin-3a(-)在AdaBoost+SVM 和其他模型中的ROC曲线Fig.4 ROC curves of Nutlin-3a(-)in AdaBoost+SVM and other models
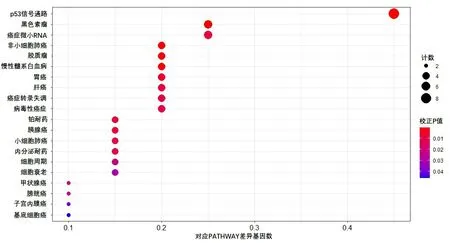
图5 Nutlin-3a(-)药物集重要特征的KEGG富集结果Fig.5 KEGG enrichment result of important characteristics of Nutlin-3A(-)drug set
本研究的不足之处在于目前的模型只有基因表达数据作为输入特征,虽然基于单一组学进行疾病研究已经发现了诸多新的致病因子,但疾病的发生发展是一个复杂的网络,基因拷贝数变异、体细胞突变、甲基化的异常等诸多因素都会影响生命体特征和药物反应[17]。因此,之后的研究会处理以上多组学数据作为训练特征,同时尝试更多方法建模,以达到更好的肿瘤药物预测效果。
猜你喜欢
现代仪器与医疗(2022年4期)2022-10-08
电气技术(2022年6期)2022-06-27
电子产品世界(2022年4期)2022-04-21
科学与生活(2021年16期)2021-11-25
计算机系统应用(2021年2期)2021-02-23
现代临床医学(2021年1期)2021-01-26
甘肃教育(2020年4期)2020-09-11
电子技术与软件工程(2019年18期)2019-11-18
电子技术与软件工程(2017年14期)2017-09-08
医学研究杂志(2015年9期)2015-07-01
