
Dominant early heading without yield drag in a sister-line BC breeding progeny DEH_229 is controlled by multiple genetic factors with maineffect loci
2021-05-06 13:02:54MuhiuddinFruqueeQingZhngLubioZhngLinyunXuWenshengWngJinsnChenJinlongXuZhikngLiBinyingFuTinqingZheng
The Crop Journal 2021年2期
Muhiuddin Fruquee , Qing Zhng , Lubio Zhng , Linyun Xu ,Wensheng Wng , Jinsn Chen , Jinlong Xu ,b, Zhikng Li ,b, Binying Fu ,b,,Tinqing Zheng ,
a Institute of Crop Sciences/National Key Facility for Crop Gene Resources and Genetic Improvement, Chinese Academy of Agricultural Sciences, Beijing 100081, China
b Shenzhen Institute of Breeding & Innovation, Chinese Academy of Agricultural Sciences, Shenzhen 518120, Guangdong, China
c Institute of Rice Research, Guangdong Academy of Agricultural Sciences, Guangzhou 510640, Guangdong, China
Keywords:
ABSTRACT Dominant early heading (DEH) in rice (Oryza sativa L.) is of interest in both breeding and genetics.The genetic mechanisms underlying DEH have remained largely unclear.We have developed a near-isogenic DEH line without yield drag named DEH_229 by sister-line backcross (BC) breeding with MH63, a restorer, as the genetic background.We conducted a pilot genetic investigation under both short-day (SD) and long-day (LD) conditions.The DEH line harbored only 1.06% variation in the genome sequence relative to MH63.The variants were distributed throughout the genome.Using QTL mapping by sequencing (QTL-seq) on an F2 population derived from a cross of MH63×DEH_229, 57 loci were detected under the SD condition.Joint mapping employing a genome-wide association study with accessions from the 3000 rice genome sequencing project (3K-RG), reduced the number of QTL by 43.9%.Using Rice Functional Genomics & Breeding (RFGB) database, the number of SNP cluster regions within the QTL regions reduced by 27.3%.Further comparison of the genome variation between DEH_229 and MH63 in addition to gene annotation information revealed a new DEH allele of DTH3 with multiple variable sites as a possible major factor underlying the early-heading phenotype of DEH_229.An InDel marker, ZMEH_1, was designed based on the variation between DEH_229 and MH63 within this region.It accounted for 86.0% of heading date variation under both SD and LD conditions in 109 randomly chosen progeny derived from extreme lines of the MH63×DEH_229 population.This study reveals the genetic complexity of DEH in the near-isogenic line and may provide useful material and marker information for plant molecular breeding.
1.Introduction
Rice (Oryza sativa L.) is a model species for crop research.Flowering time or heading date (HD) shapes variation among cultivars.As rice is also a major human food source worldwide, its grain yield (GY) is a main target trait of breeding.Thus, knowledge of pleiotropic genes for both HD and GY would be of great value.However, for most reported pleiotropic genes, increased GY is linked with delayed HD [1-3], hindering their direct application in cultivar development.
Lines showing dominant early heading (DEH) have long been known or have been developed.A partial DEH mutant was found in the same mutagenized population that produced Calose 76 with sd1 [4].Two DEH mutants, R1-8 and R1-2, were derived from IR20 [5].An early line 6442S-7 selected from the male-sterile line 6442S showed complete DEH with no yield drag [6].The DEH in Kefeng A was found to vary by cross [7].Both Zaoxian A [8]and H14 [9]are progeny of wide crosses and display partial DEH.The DEH of D64B, an indica maintainer, is almost complete and is not affected by day length, especially in crosses with lines that are genetically distant [10].Another indica maintainer, Lexiang 202B, showed partial DEH [11].A DEH sterile line, UP-3s, was crossed with late restorer lines to obtain earlier-maturing hybrids to avoid the high-temperature period in Arkansas, USA [12].Most DEH lines are sterile/maintainer lines.
Although HD is a highly heritable trait in rice, genetic analysis of DEH often takes a long time because of the multiple complex factors that underlie it.For example, Ef-1, a locus that was first mapped on chromosome 10 [13], was later discovered to harbor multiple alleles [14]and was allelic to Ehd1.It promotes flowering especially under short-day (SD)conditions independent of Hd1 [15]and functions in a homodimer structure [16].H14 carries Ef-h(t) on chromosome 6 [9].Kefeng A carries both Ef-1 and a dominant suppressing gene for E1 (Su-E1) together with a recessive suppressor gene for Se-1 (i-Se-1) [7].In Le-Xiang202B, the major genetic factor, Ef-2(t) [11], may be allelic to Ef-3(t) on chromosome 2 in D64B[10].The DEH of Zaoxian A was controlled by a partially dominant gene [17,18].In the first mapping report [6], 6442S-7 was found to harbor a pair of major genes controlling its DEH.After about 20 years, one of them named Ef-cd (Early Flowering - Complete Dominant) has been recently identified [19].
The genetic mechanisms underlying DEH have remained unclear.We previously developed a new DEH line without yield drag in a restorer genetic background.We describe a pilot genetic investigation of DEH_229 intended to elucidate the complexity of DEH and generate useful material and information for designed breeding early-maturing cultivars.
2.Materials and methods
2.1.Plant materials
A line with early-heading character under both long-day (LD)and SD conditions, DEH_229, was derived from a cross between sister lines, MH63 and 3027, in 2012 (Figs.S1 and S2).To identify the genetic bases of the early heading character of DEH_229, a single cross was made using MH63 as female and DEH_229 as male parent.Two elite restorer lines, 9311 and R498, were also used as female parents in test crosses with DEH_229.Seeds from these F1plants with a uniform early-heading phenotype were bag-harvested.An F2population of 6705 plants(Table S1) was derived from a cross of MH63×DEH_229 and the two parents and F1plants were tested.
2.2.Phenotyping and genotyping
Since 2016, the flowering characteristics of DEH_229 and MH63 have been continuously observed under both LD (major season) and SD (winter season) conditions.The planting locations were Changping station (Beijing, 40.17°N, 116.23°E, for LD condition) and Hainan station (Sanya, 18.30°N, 109.30°E,for SD condition) of Institute of Crop Sciences, Chinese Academy of Agricultural Sciences (ICS, CAAS).
Based on observation of the parents (Fig.S1), an F2mapping population derived from the MH63×DEH_229 cross was tested under SD conditions at Hainan station for phenotyping during the winter season of 2017.The HD phenotype of the MH63×DEH_229 F2mapping population was used for selection of representative plants from the two extreme (early and late) pools and their seed was advanced to the F3generation.F3families were then planted at Hainan again during the winter season of 2018 and non-segregating lines in HD were visually selected under SD conditions.Seeds from these lines were then sown at Beijing in 2019 for evaluating the HD phenotype under LD conditions.The testing generation of these lines was F4.From the F4lines, which behaved consistently with their F2ancestors with respect to the HD phenotype(early or late), a random sample was chosen for later confirmation of an InDel marker.The F1and F2progenies derived from 9311 and R498 crossed with DEH_229 were also planted during the winter season of 2018.MH63 and DEH_229 were used as the controls in all seasons.Field management followed standard local procedures.Days to heading (DTH) of a single plant in an F2population or a single line in the F4generation were recorded when 50% of the tillers were heading.All parents and F1progenies were planted in a randomized complete block design for field layout with at least three replications.
From the MH63×DEH_229 F2mapping population, 222 plants with the same late-heading phenotype as the female parent (MH63) were used to construct a wild-type pool (WTpool), and 454 plants with the same early-heading phenotype as the male parent (DEH_229) were used to construct a mutant-type pool (Mut-pool).All of these plants were tagged and then bag-harvested separately.Approximately 15 seeds from each individual were germinated for leaf sampling at the three-leaf stage.Leaf samples of equal weight from each individual that belonged to the same pool were then bulked.The bulked leaf samples from the WT- and Mut-pools,together with the female and male parents, were subjected to genomic DNA extraction using the CTAB method.Libraries with peak insert sizes of about 200 bp were prepared using an Illumina Truseq DNA library protocol (Illumina Kit FC-121-4001; Illumina Inc., San Diego, CA, USA).Library quality was checked using the standard protocol of the Agilent 2100 Bioanalyzer High Sensitivity Kit [20].After library profile analysis,the libraries were sequenced by next-generation sequencing(NGS) using the 150-base paired-end strategy in the Illumina HiSeq X10 platform (Illumina).The two original parents,3027 and MH63, were also assayed using respectively the 50K SNP chip and 451 SSRs, to identify their genomic variants,as previously described [21].
Based on the four InDel among six variations around DTH3 between MH63 and DEH_229, eight InDel markers were designed using AmplifX [22].The PCR amplification was performed according to the routine protocols adopted in the Rice Molecular Breeding Lab of ICS, CAAS with the following parameters: an initial denaturation at 95 °C for 3 min, 40 cycles of 95 °C for 30 s, 57 °C annealing for 30 s, and 72 °C extension for 35 s, with a final extension of 5 min at 72 °C.About 1.2 μL of PCR product from each sample was separated by 8% polyacrylamide gel electrophoresis.
2.3.Data analysis
Raw reads from NGS were screened by quality check [23].All reads passing the quality check were then aligned against the reference map of IRGSP-1.0 [24].Genome variations were called with GATK [25]and stored in standard variant call format (VCF).Perl scripts were used to extract SNP data from the VCF files for quantitative trait locus (QTL) mapping.Mapping was performed with the QTLseqr package [26].
A joint-mapping procedure was applied as previously[27,28]described with minor revisions based on the outputs from QTLseqr, and GWAS was performed using HD data collected at Hainan in the same season for 3K-RG [29].Loci identified by both QTLseqr in the F2population and GWAS in 3KRG were then subjected to SNP-cluster screening with the Haplotype module in RFGB v2.0 [30].Another module of RFGB(SNP & InDel) was used for further screening of candidate genes in regions harboring candidate SNP clusters from the previous step.During the primer design for confirmation,the reference map of MH63RS2 [31]was adopted.Common statistical analyses including Chi-square test were performed with Microsoft Excel [32].
3.Results
3.1.Phenotypic characters of DEH_229 and related materials
The early heading line, DEH_229, was derived from consecutive backcrossing using MH63 as the recurrent parent and a sister line, 3027, as the donor (Fig.S1).MH63 and 3027 had similar phenotypes, including flowering time (Fig.S2).However, DEH_229 differed markedly from MH63, especially inflowering time with no yield drag (Fig.1a, b; Fig.S1).The F1plants produced using restorers MH63, 9311, and R498 as female parent (P1) and DEH_229 as male parent (P2) showed both earlier maturity and higher GY than either parent (Figs.1 and 2).Other yield traits varied with the cross.In the cross with MH63, panicle number (PN) and spike fertility (SF) were significantly increased in F1plants compared with both MH63 and DEH_229.Significant heterosis of GY was present,with no significant influence on grain shape (Fig.1c).In the 9311 cross (Fig.2a, c), the GY of F1plants was also significantly higher than that of either parent.The other phenotypes of F1plants were intermediate between 9311 and DEH_229, but the trends varied by trait.Plant height (PH) and PN seemed to be more inclined to DEH_229, whereas SF, grain number per panicle (GNP) and thousand grain weight (TGW) were more inclined to P1.In the R498 cross, similar patterns were observed (Fig.2b, d).
The DTH of the F2plants derived from the cross between MH63 and DEH_229 together with the F1plants and their parents were recorded under the SD conditions of Hainan.A bimodal distribution was found for DTH in the F2population(Fig.1d).According to the distribution, the DTH values of both F1and the major part of the F2fell into the range of DEH_229.In contrast, the DTH values of the minor part of the F2overlapped the value range of MH63.However, the ratio of early(DEH_229 type) to late (MH63 type) was sharply divergent from 3:1 (5441/1264; χ2= 135.2, χ2(0.05,1)= 3.84).
Progenies from the 120 extreme F4lines behaving consistently in HD under both SD and LD conditions were considered for inclusion in materials selected for marker confirmation (Table S1).Among them, 11 late lines failed to set seed at Beijing in 2019.Finally, 109 lines (60 early and 49 late) in F4generation were subjected to further marker assay.
3.2.Genome variation in DEH_229 and related materials
Genome sequencing yielded 166.5 million clean reads for both parental lines and 143.9 million clean reads for the two pools.Most (98.4%) of these reads were mapped to the IRGSP-1.0 reference genome, resulting in mean depths of 35.7× for both parents and 30.8× for the two pools (Table S2).
High genome similarity (98.8%, 50K SNP; 97.8%, 451 SSR)was detected between the sister lines MH63 and 3027, which were the original parents of DEH_229.The variants along the genome were clustered mainly on chromosomes 5, 6, 7, 9, and 11.The overall genomic difference between DEH_229 and MH63 was estimated as only 1.06% based on the variants identified by sequencing (results not shown); however, the SNP variation between DEH_229 and MH63 was distributed throughout the genome and not just on the five chromosomes(Fig.3).
3.3.Joint mapping of loci controlling heading date
A total of 57 loci were detected by QTLseqr at a threshold of about 3.9 (Fig.3a; Table S3).Only 25 (61.4%) of them matched loci detected using the 3K-RG GWAS (Fig.3b; Table 1).Loci with the top three G′values were qEH_3a (10.1), qEH_3b (9.7),and qEH_5c (11.8); among them, qEH_3a also had the highest-log10(P) value (6.7).Distributions of SNP and InDel variants between DEH_229 and MH63 compared to the QTL distributions are presented in Fig.3.The distribution of dominant early-heading QTL loci along the genome did not closely correspond with the distribution of genome variations.
Comparative mapping revealed that eight (32.0%) of the 25 QTL regions overlapped known genes associated with flowering time in rice (Table 1).Among them, qEH_3a overlapped five known genes: Os03g0112700 [34], Os03g0122600 [37],Os03g0131900 [38], Os03g0151300 [39], and Os03g0169600 [40].Three of the EH loci overlapped two known genes: qEH_2b overlapped with Os02g0152500 [34]and Os02g0152900 [35],qEH_5c overlapped Os05g0595300 [47]and Os05g0597100 [48],and qEH_9a overlapped Os09g0240200 [42]and Os09g0134500[43].The other four loci overlapped single known genes:qEH_1b (Os01g0385400 [33]), qEH_3b (Os03g0719800 [41]),qEH_5b (Os05g0343400 [49]), and qEH_9c (Os09g0507200 [44]).
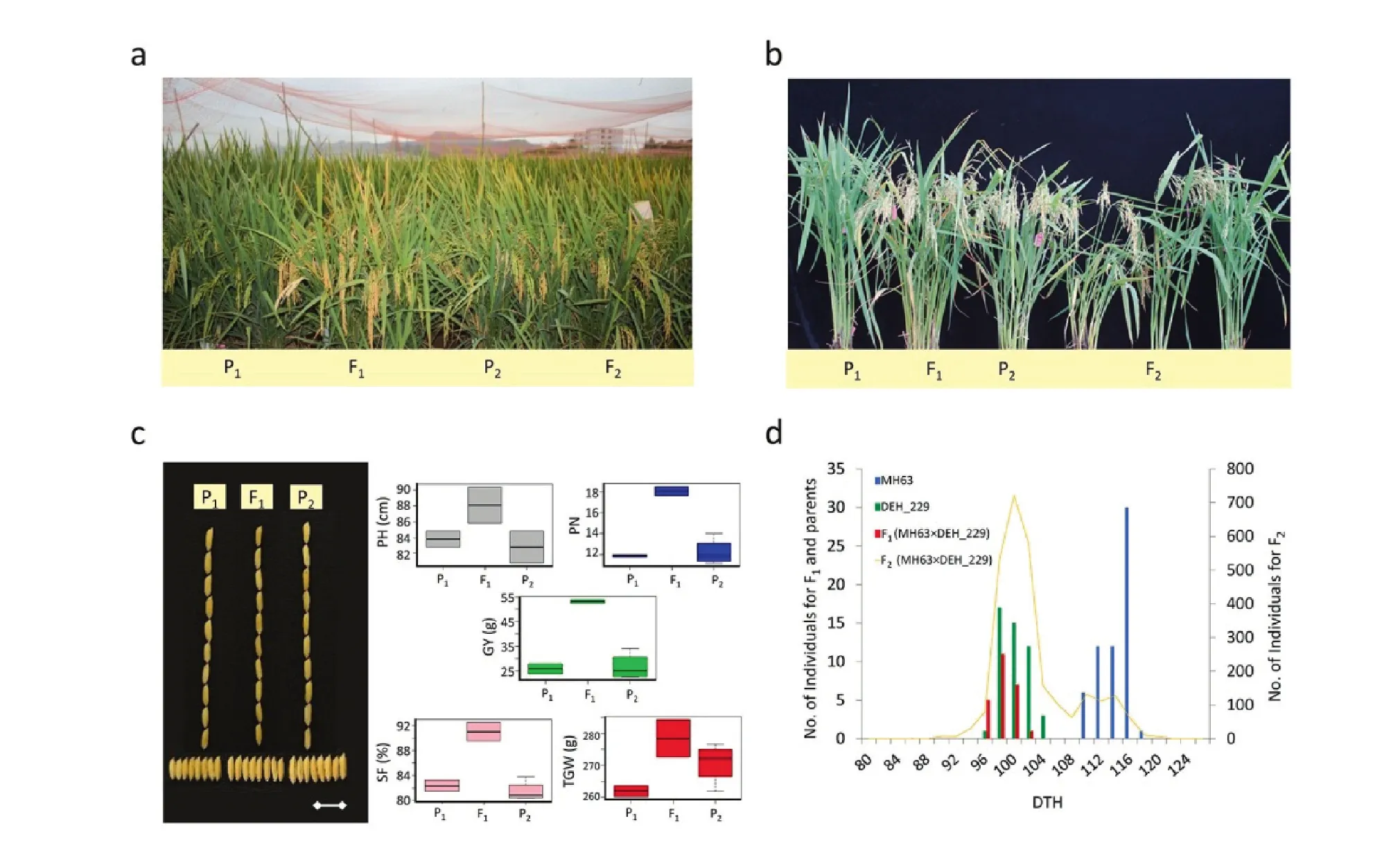
Fig.1–Phenotypes of MH63, DEH_229 and their progenies.a) and b) show phenotypes of representative individuals in field and pots respectively.c) Comparison of plant height (PH), panicle number (PN), spike fertility (SF), thousand-grain weight(TGW), and grain yield (GY) in addition to the grain shapes (Scale bar, 1 cm) between parents and F1 progeny.d) Distribution of days to heading (DTH) in parents and progenies.P1, MH63; P2, DEH_229; F1, (MH63×DEH_229) F1; F2, (MH63×DEH_229) F2.
3.4.Joint screening with RFGB v2.0
For further analysis, 44 SNP clusters in the 25 QTL regions were grouped according to an empirical threshold of approximately 200 kb (Table 2).Haplotype analysis with the SNP sites in RFGB v2.0 revealed that 32 (73%) of the clusters were significantly associated with HD in 3K-RG.Among the 32 SNP clusters significantly associated with HD, one region, EH_9 in qEH_3a, was supported by both QTLseqr and GWAS (Table 1)and by the gene annotation information from the SNP & InDel module of RFGB v2.0 [30].
In EH_9, six candidate genes were associated with HD(Table 3).Haplotype variations in all genomic regions of these genes were significantly associated with DTH in the 3K-RG.Among them, all the 500bp_prom regions were significant.However, the coding sequence (CDS) regions were significant in only three genes (Os03g0122600 [37], Os03g0151300 [39], and Os03g0169600 [40]).The 3′ UTR and 2-kb promoter regions were significant in two genes (Os03g0112700 [36]and Os03g0131900 [38]).All the other regions were significant in another two genes (Os03g0122600 and Os03g0169600).
Finally, of the variants between DEH_229 and MH63, only the Os03g0122600 genomic region harbored variants (two SNPs and four InDels, Table 3).The other five candidate genes were monomorphic between DEH_229 and MH63 at the genomic sequence level.Os03g0122600 (Table S4; Fig.4), which is located between 1.14 and 1.27 Mb in EH_9, was associated with flowering time in rice under both LD and SD conditions[37].In the CDS region of Os03g0122600, three haplotypes were identified, among which Hap14 showed the shortest DTH (Fig.4a).In the promoter region, nine haplotypes were found in 3K-RG, among which Hap1, Hap3, and Hap8 represented the shortest DTH (Fig.4b).Both 5′UTR and 3′UTR contained three haplotypes.In both 5′UTR and 3′UTR, Hap2 showed the shortest DTH (Fig.4c, d).
3.5.Confirmation of a candidate of EH9 with marker assay
For molecular breeding application, we used markers to evaluate the contribution of variants in Os03g0122600 to the earlyheading phenotype of DEH_229.The 109 F4lines (60 early and 49 late) derived from the selection procedure were assayed with InDel markers in the target region.
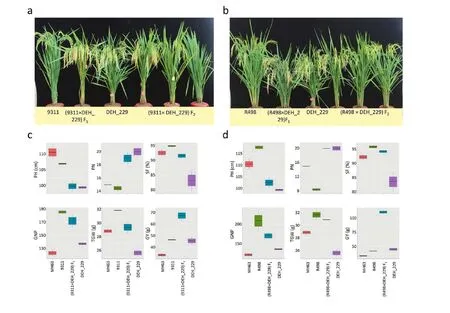
Fig.2–Phenotypic effects of DEH_229 on representative restorer lines 9311 and R498.a) Phenotypes of 9311, DEH_229, and their progenies.b) Phenotypes of R498, DEH_229, and their progenies.c) Comparisons of plant height (PH), panicle number(PN), spike fertility (SF), grain number per panicle (GNP), thousand-grain weight (TGW), and grain yield (GY) among MH63,9311, DEH_229, and the (9311×DEH_229) F1 generation.d) Comparisons in PH, PN, SF, GNP), TGW, and GY among MH63, R498,DEH_229, and the (R498×DEH_229) F1 generation.
PCR primers were designed according to the four InDel variants between DEH_229 and MH63 with Var_SN of 2, 4, 5,and 6 (Table S4; Fig.5).Among the eight primer pairs designed(Table S5), three were polymorphic between the parents.Among them, ZMEH_1 presented the best resolution and accounted for 86.0% of phenotypic variation in 109 F4progeny of the MH63×DEH_229 population under the LD condition(Fig.5).
4.Discussion
4.1.Using multiple tools to narrow a set of loci
We previously [27]employed joint mapping using RIL population linkage and GWAS following an approach first described in maize (Zea mays L.) [28].NGS-Bulk Segregant Analysis(NGS-BSA) is a rapid method for identifying QTL.In our previous study [27], joint mapping of leaf rolling loci effectively reduced the target loci from 14 to five.In the present study,our target loci were reduced from 57 to 25 (a >56% reduction)by joint use of both the QTLseqr toolkit for NGS-BSA [26]and 3K-RG GWAS.Critical mutations of agronomically important genes in rice are commonly rare mutations [50,51].The SNP data for 3K-RG used in our GWAS were filtered by screening for major allele frequency (MAF) > 0.05 [29], which may have largely ruled out critical rare mutations.This may explain why qEH3a, the second highest peak in the mapping results from QTLseqr, was identified rather than the top 1 peak on chromosome 1 [26].
Within the 25 loci, 44 SNP clusters (EH_1-EH_44) remained.We used the ‘‘SNP site” function embedded in the Haplotype module of RFGB.Screening out the 27.3% of insignificant clusters prior to additional lab work saved materials and labor.
4.2.Genetic bases for dominant early maturity in DEH_229
Known dominant early heading genes or QTL are located on chromosomes 2, 3, 8, and 10 [52].Our mapping results showed that related loci in DEH_229 were distributed all over the genome, even after joint screening by 3K-RG GWAS.
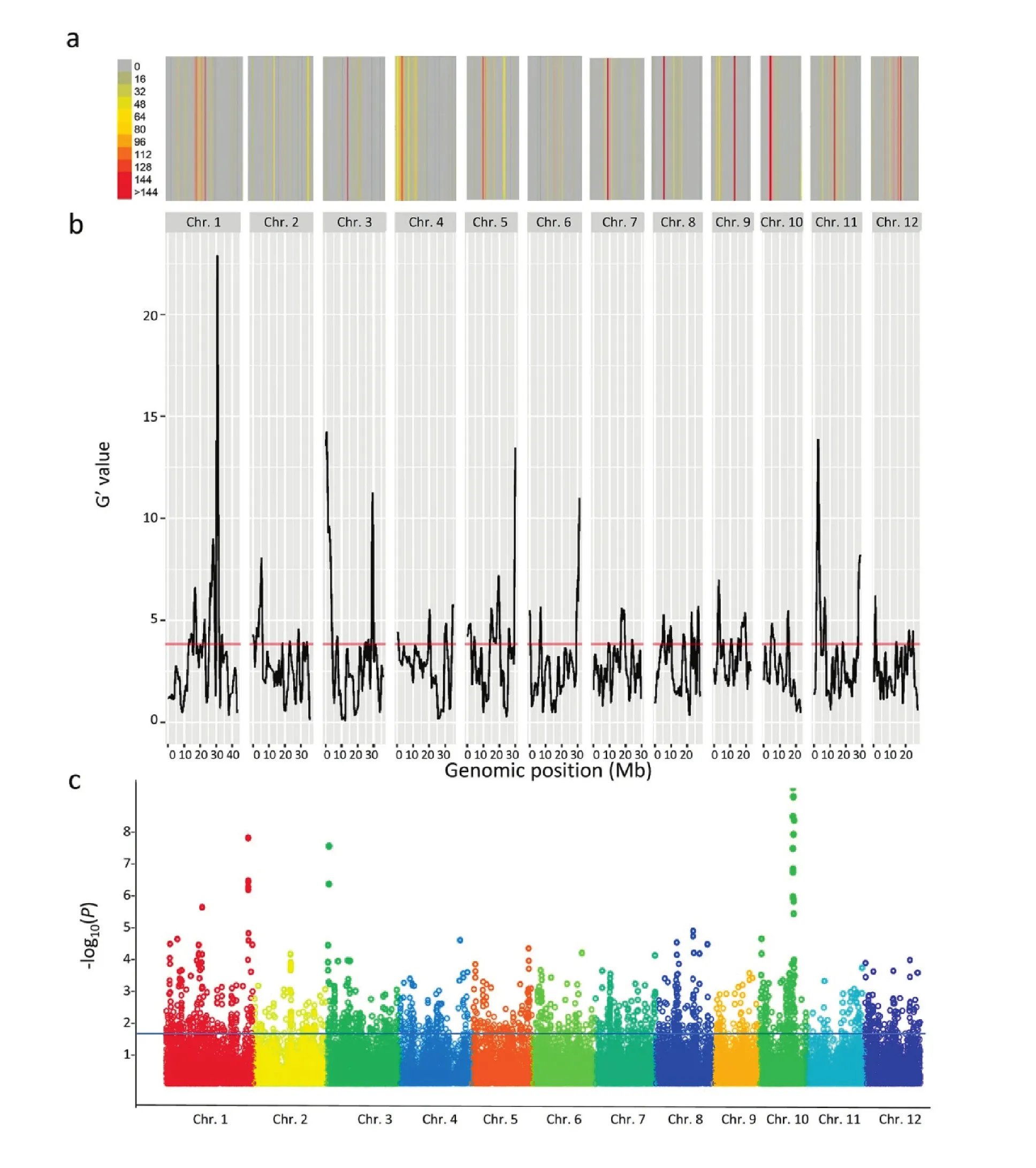
Fig.3–A genome view of variation and flowering-time associated loci.a) Number of variants (SNP and InDel) between DEH_229 and MH63.b) G′ values detected by QTLseqr [26]in MH63×DEH_229 F2 population.c) Manhattan plots for flowering variation in 3K-RG.
The Ef-cd locus was identified as an lncRNA [19], which promoted flowering by upregulating DTH3/OsSOC1/Os03g0122600 expression.The medium-late restorer 9311 is known to carry a dominant suppressor gene (Su-Ef-cd) that can partially suppress the dominant early flowering genes under SD conditions, including Ef-cd, which is carried by 6442S-7 [53].However, DEH_229 was still functioning in the 9311-cross under SD conditions.This finding strongly suggested that there would be genetic mechanisms other than Ef-cd underlying DEH_229.With the aid of genomic sequence analysis, the genomic variants between DEH_229 and its recurrent parent MH63 were localized to around Os03g0122600 (DTH3) rather than Os03g0122500 (Ef-cd).Interestingly, Os03g0122600 is a target gene regulated by Ef-cd in 6442S-7 [19].With the aid of marker ZMEH_1, the phenotypic contribution of the DEH_229 allele around DTH3 was further confirmed (Fig.5).

Table 1–QTL loci jointly detected by QTLseqr and 3K-RG GWAS.
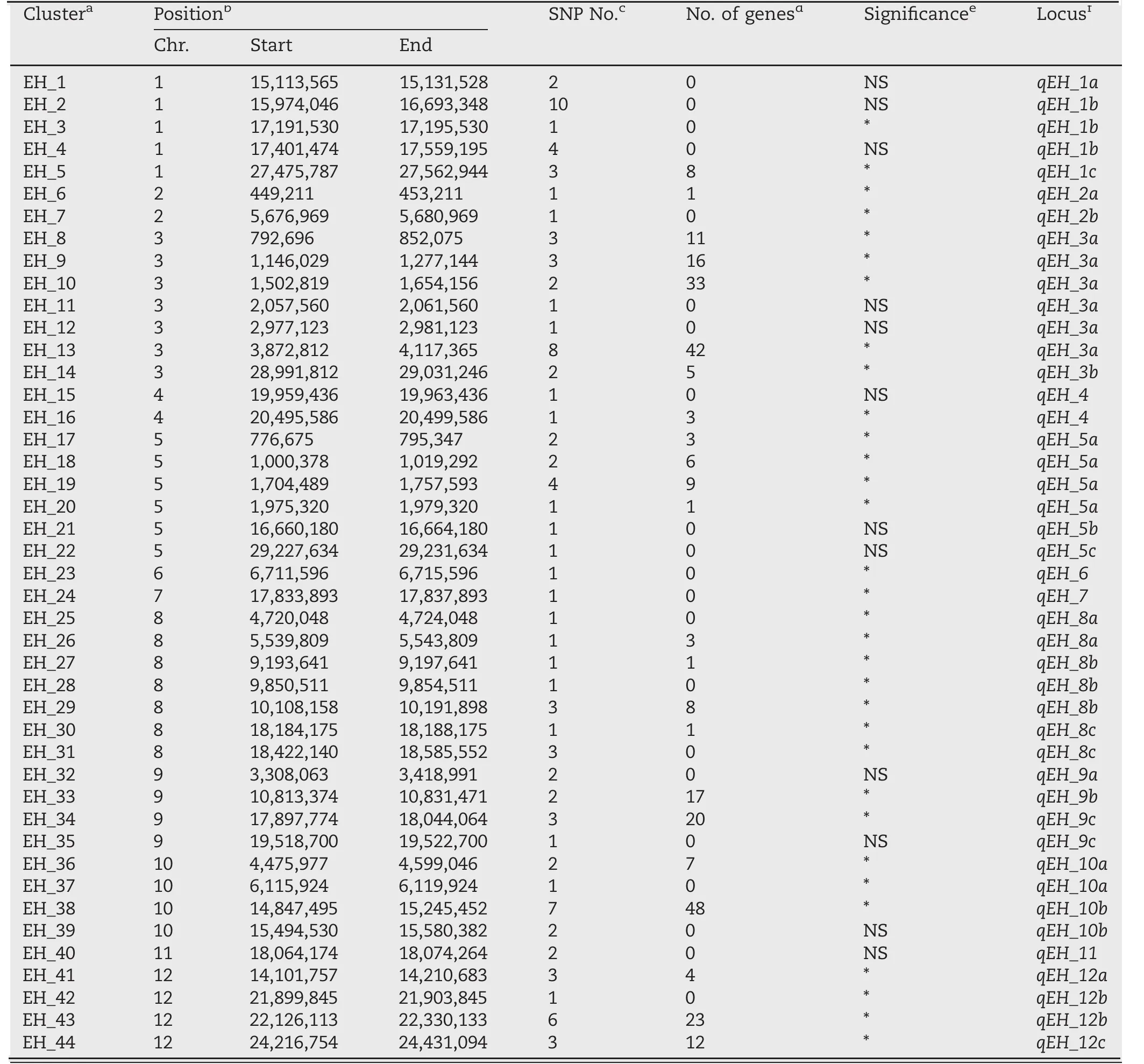
Table 2–Candidate clusters for SNP-based haplotype analysis using RFGB.
Genomic differences around the DTH3 region were also found between reference maps, IRGSP1.0 and MH63RS2.In IRGSP1.0, all six variants were located within the genomic region (Table S4).However, in MH63RS2, most (Var_SN 1-5)of the six variants, in particular Var_SN 2, were located in intergenic regions, where no gene has been annotated(Fig.5).Os03g0122600 (DTH3, LOC_Os03g03100) has no corresponding annotation in MH63RS2, as is also the case for OsMH_03G0019100 in IRGSP1.0 according to the RIGW database [54].This result suggests that complex molecular mechanisms, rather than a single new allele of DTH3, contributed to the dominant early heading phenotypic variation of DEH_229.
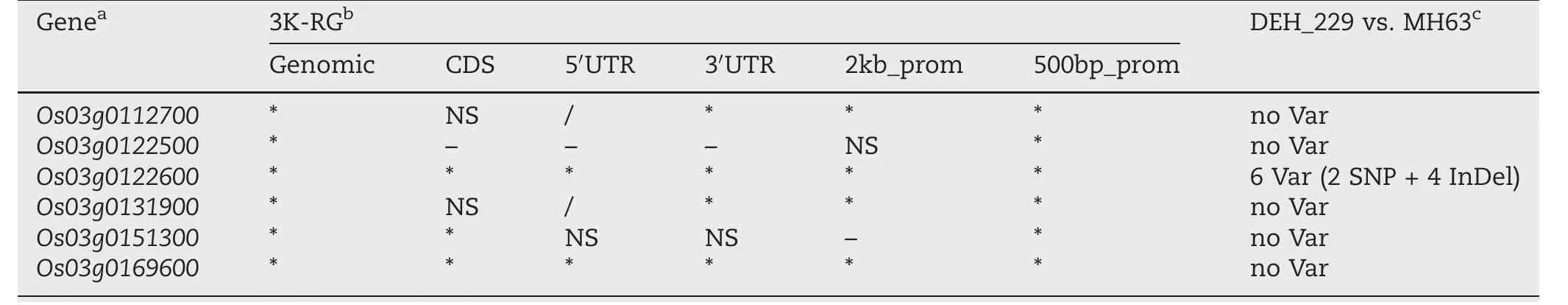
Table 3–Variants in gene regions in EH_9 using HD data from RFGB and comparison between DEH_229 and MH63.
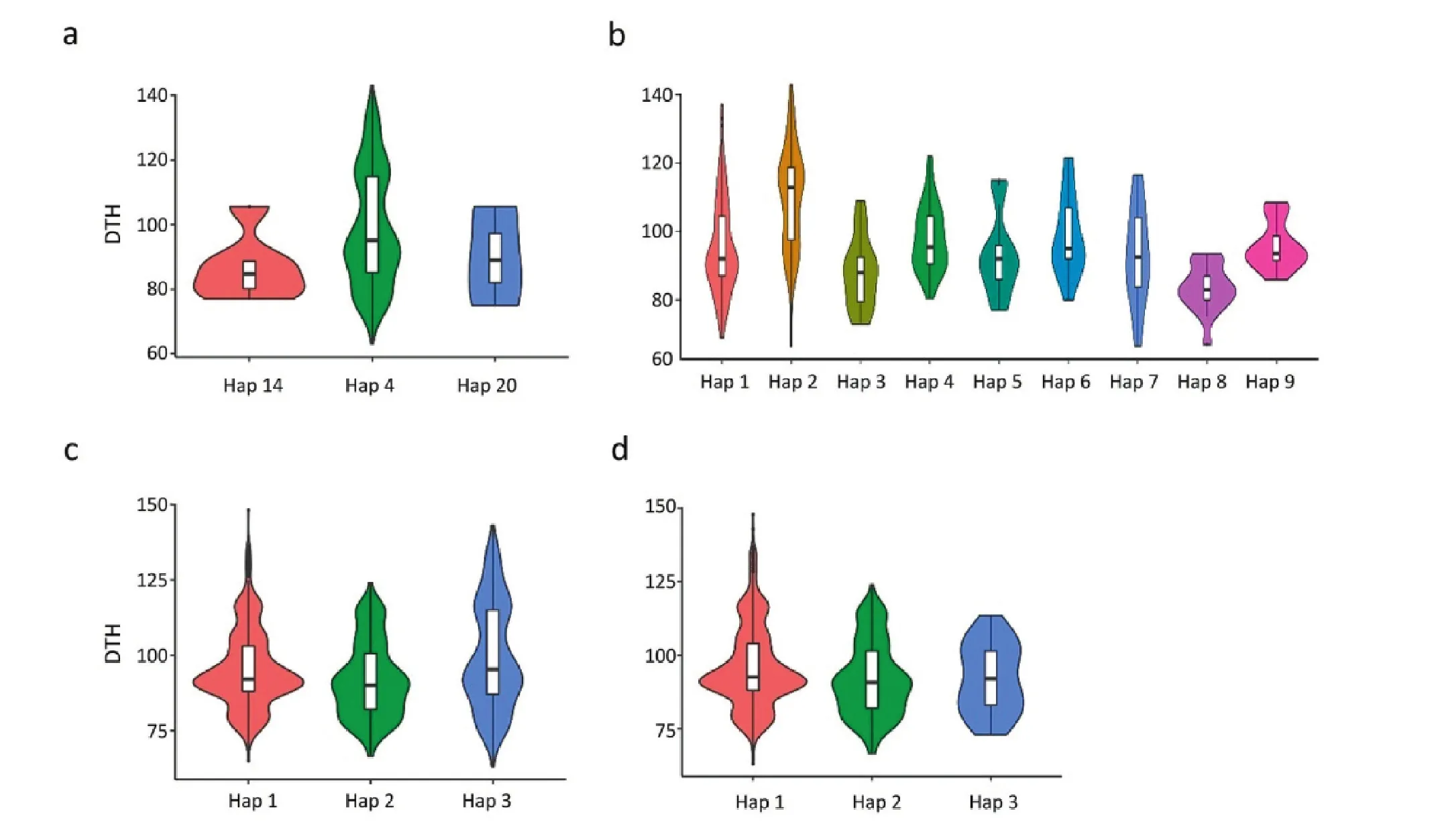
Fig.4–Violin plots for the haplotype analysis results of one candidate gene Os03g0122600.Phenotypic effects of haplotypes in the regions of a) CDS, b) 2-kb promoter, c) 5′UTR, and d) 3′UTR.
4.3.Possible application of DEH_229
Improvement of adaptability and grain yield are two of the most vital goals of crop researches.Breeding materials derived from DEH without yield drag are of great values.Through a stepwise series of crosses, early-maturing and semi-dwarf mutations were recombined with a gene for glabrous hull to produce the early-maturing, semi-dwarf,glabrous-hull cultivar M101 [55].Ef-cd has been introduced into a medium-late-maturing japonica cultivar, Koshihikari,to produce early-maturing elite lines with improved adaptation to cultivation in Jilin province in northeast China (Fang Jun, personal communication).In the test crosses with MH63, R498, and 9311, the possible breeding value of DEH_229 was strongly indicated.Further tester materials and in particular sterile lines could be evaluated for breeding schemes involving DEH_229.
Improved yield potential of hybrids is largely achieved by high yield potential in parents [56].In indica hybrid breeding,9311 and R498 belong to an advanced generation of restorer line products; they have higher GY than MH63 in terms of biomass (Fig.2c-d).The F1plants produced by crossing the MH63×DEH_229 genetic background with 9311 and R498 showed highly increased GY compared to the parents.Thisfinding strongly suggests that DEH_229 will provide useful breeding materials for indica restorer improvement, especially in shortening maturity duration without yield drag.The InDel marker ZMEH_1 may help in following the early-HD phenotype of DEH_229.Moreover, crossing with DEH_229 can produce F1plants with both early-maturity phenotypes similar to DEH_229 and high GY, even using MH63, which harbors only 1.06% of the associated genomic variation found in DEH_229.DEH_229 would also likely offer additional useful material for genetic analysis of heterosis.

Fig.5–Marker confirmation for one candidate gene Os03g0122600 in the F4 population of MH63×DEH_229.a) Positions of the six variants between MH63 and DEH_229 according to the reference map of MH63RS2.b) Gel assay by InDel marker ZMEH_1.Lanes 3 and 65: MH63; lanes 4 and 66: DEH_229; lanes 5–64: representative lines with consistency between phenotype and marker genotypes, lanes 5–33: lines with early heading date as DEH_229, and lanes 34–64: lines with late heading date as MH63; lanes 1, 2, 67, and 68: ladders.
DEH_229 was created from a cross between sister lines 3027 and MH63 via backcrossing breeding (Fig.S1).It showed early flowering in comparison with MH63 under both SD and LD conditions.The elite phenotype was exposed by crossing and selfing rather than through parental heritage.This finding is evidence of cryptic genetic variation (CGV) [57]or ‘hidden diversity” [58].CGV may be used to develop new nonparental phenotypes [59].CGV-like phenomena in DEH_229 await future investigation.
Declaration of competing interest
Authors declare that there are no conflicts of interest.
Acknowledgments
We appreciate supports from the National Key Research and Development Program of China (2016YFD0101801), the National Natural Science Foundation of China (31871715),the Agricultural Science and Technology Innovation Program of Chinese Academy of Agricultural Sciences (ICS2020YJ07BX,1610092015003-10, and Y2020PT24), the Open Program from the Guangxi Key Laboratory of Rice Genetics and Breeding(2018-05-Z06-KF01), and the ‘‘Green Super Rice” Project from Bill & Melinda Gates’ Foundation (OPP1130530).We also thank Dr.Yongming Gao for providing restorer lines including R498 and 9311, Dr.Chun-Chao Wang for aid in raw read cleaning.
Author contributions
Tianqing Zheng and Binying Fu conceived and designed the experiments.Muhiuddin Faruquee, Qiang Zhang, Lubiao Zhang, Linyun Xu, and Tianqing Zheng performed the experiments.Tianqing Zheng and Muhiuddin Faruquee analyzed the data.Wensheng Wang, Jiansan Chen, Jianlong Xu, and Zhikang Li contributed reagents/materials/analysis tools.Tianqing Zheng and Muhiuddin Faruquee wrote the paper.
Appendix A.Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2020.06.014.

- The Crop Journal的其它文章
- Brief Guide for Authors
- Short Communication Seed-specific overexpression of cotton GhDGAT1 gene leads to increased oil accumulation in cottonseed
- Waterlogging stress in cotton:Damage,adaptability,alleviation strategies,and mechanisms
- Genome-wide alternative splicing variation and its potential contribution to maize immature-ear heterosis
- Identification of microRNAs involved in crosstalk between nitrogen,phosphorus and potassiumunder multiple nutrient deficiency in sorghum
- RNA interference targeting ω-secalin genesdifferentially affects the processing quality in a wheat T1BL·1RS translocation line