
7H-双呋咱并[3,4-b∶3′,4′-f]氧化呋咱并[3″,4″-d]氮杂环庚三烯新法合成、晶体结构与性能
2021-05-06 07:36:28翟连杰罗义芬李祥志杨凯迪毕福强陈三平王伯周
火炸药学报 2021年2期
翟连杰,罗义芬,李祥志,杨凯迪,毕福强,陈三平,王伯周
(1.西安近代化学研究所,陕西 西安 710065;2.氟氮化工资源高效开发与利用国家重点实验室, 陕西 西安 710065;3.西北大学 化学与材料科学学院,陕西 西安 710127)
引 言
硝基是含能化合物中具有重要作用的基团[1-3]。一方面,在CHON有机分子中引入硝基是提升含能化合物密度的有效手段;另一方面,硝基也是分子内“氧”的来源,在爆轰反应中,硝基的氧将C、H等氧化成氧化态,进而释放能量。然而,在向单个分子结构中引入更多数量硝基时,尽管密度和爆轰性能在一定程度上得到提升,但也会导致目标化合物合成步骤长、合成难度大、制造成本和感度高、分解温度低等问题,严重制约其在武器装备中的应用。
氮杂芳环(吡唑、咪唑、三唑、噁二唑、四唑、四嗪等)因其优异的理化性能和爆轰性能在炸药、推进剂和发射药领域显现巨大潜力[4-6]。与传统含能材料相比,氮杂芳环属于共轭体系,具有热稳定性好和感度低的特点,同时分子内含有丰富的N—N和C—N键,因而具有高的正生成焓和密度[7-8]。尤其是呋咱类含能化合物分子内含有两个N—O键,其作用相当于硝基,为分子提供活性“氧”,有效改善氧平衡差的问题,受到世界各国含能材料研究者的高度关注[9-11]。
近年研究结果表明[12-13],氮杂芳环构成的无硝基平面结构化合物普遍具有较高的晶体密度,已成为提升含能化合物密度的有效策略。完全共平面结构分子,分子间存在较强π-π作用,能够有效拉近分子之间的距离,形成层状排列,实现分子之间更加紧密的堆积,表现出更高的密度。氮杂芳环上的N—O键能够起到硝基作用,并在一定程度上实现降低感度的目的。俄罗斯科研人员[14-16]于1995年合成了呋咱并[3,4-e]-1,2,3,4-四嗪-4,6-二氧化物(FTDO),其密度为1.85g/cm3,生成焓约为652.7kJ/mol,理论爆速和爆压分别为9802m/s和44.8GPa。2014年美国爱达荷大学Shreeve J M课题组[17]以二氨基呋咱为原料,经缩合环化、取代、氨化和氧化反应合成了1,2,3-三唑并[4,5-e]呋咱并[3,4-b]氧化吡嗪,该化合物密度为1.85g/cm3,爆速和爆压分别为8532m/s和32.4GPa,撞击感度高达32J,是一种潜在的富氮钝感高能炸药。国内王伯周等[18]合成了双呋咱并[3,4-b∶3′,4-f]氧化呋咱并[3″,4″-d]氧杂环庚三烯(BFFO),其晶体密度为1.87g/cm3,熔点为92~94℃,实测爆速为8256m/s(压药密度为1.85g/cm3),撞击感度为12%,摩擦感度为0,特性落高为57.5cm,具有熔点低、感度低、能量密度高的特点, 在熔铸炸药中有一定的应用前景。
鉴于BFFO独特的平面环状结构和优异的综合性能,本课题组致力于7-羟基双呋咱并[3,4-b∶3′,4′-f]氧化呋咱并[3″,4″-d]氮杂环庚三烯(HATFO)的合成研究[6],尝试将3,4-双(4′-硝基呋咱-3′-基)氧化呋咱与羟胺溶液反应时,却意外得到7H-双呋咱并[3,4-b∶3′,4′-f]氧化呋咱并[3″,4″-d]氮杂环庚三烯(HATF)。尽管Stepanov A I等[19]于2012年报道了HATF的合成与部分表征数据,但其晶体数据、热性能及爆轰性能并未见报道。为此,本研究发展了一种新的HATF合成方法,虽然收率略低于文献方法,但反应机理与过程与文献方法有实质不同。结合量子化学手段完成了HATF的13C NMR和15N NMR谱归属;同时成功培养了适合单晶X-射线衍射分析的HATF单晶,获得了晶体结构数据,并采用DSC及EXPLO5程序,对其热性能以及爆轰性能进行了研究。
1 实 验
1.1 试剂与仪器
乙醚、碳酸钠、浓硫酸、30%双氧水、50%羟胺溶液、乙腈等主要试剂均为化学纯。
NEXUS870型傅里叶变换红外光谱仪,美国热电尼高力公司;AV500型(500MHz)超导核磁共振仪,瑞士Bruker公司;Vario EL-Ⅲ型元素分析仪,德国Exementar公司;LC-2010A 液相色谱仪,日本岛津公司;Q-200型差示扫描量热仪,美国TA公司;Bruker SMART APEX II CCD平面探测X射线单晶衍射仪,德国Bruker公司。
1.2 合成路线
HATF合成路线如下,其中3,4-双(4′-硝基呋咱-3′-基)氧化呋咱依据文献[20]合成。
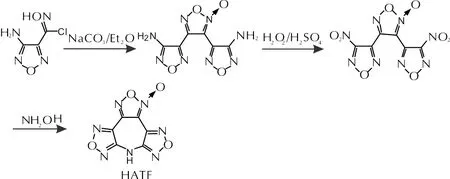
1.3 HATF的合成
室温下,将3.1g(10mmol)3,4-双(4′-硝基呋咱-3′-基)氧化呋咱加入到50mL 乙腈中,搅拌均匀后缓慢滴加0.65g (10mmol)质量分数为50%羟胺水溶液,保温反应3h,直到原料完全消失。反应结束后减压蒸馏除去乙腈,剩余物经重结晶得1.3g无色固体,收率55.3%,纯度98.2%。熔点 227.7℃, 分界点271.5℃。
1H NMR (DMSO-d6, 500MHz) ,δ: 12.56 (s, H, NH);13C NMR (DMSO-d6, 125MHz) ,δ: 152.44,151.99,145.60, 138.10, 135.83, 106.40;15N NMR (DMSO-d6),δ:-32.93,-28.58,-11.86,-11.38,-8.79, 28.05, 28.64; IR (KBr),υ(cm-1): 3293, 3215, 3113, 3080, 2976, 2817, 2705, 1655, 1633, 1615, 1572, 1553, 1492, 1462, 1443, 1393, 1359, 1225, 1152, 1062, 996, 970, 897, 879, 820, 781, 717; 元素分析(C6HN7O4, %):计算值,C 30.65, H 0.43, N 41.70; 实测值,C 30.77, H 0.39, N 40.92。
1.4 单晶培养与晶体结构测试
将纯度大于98.0%的HATF样品,加入到乙酸乙酯中,充分溶解后,得到无色溶液并置于干净的培养瓶中,室温下放置一段时间后,得到无色晶体。
选取尺寸为0.31mm×0.25mm×0.13mm的HATF单晶,室温下置于CCD衍射仪上,经过石墨单色器单色化的Mo Kα射线(λ=0.071073nm),以ω/2θ扫描方式,在2.51°≤θ≤25.1°,-5≤h≤7,-12≤k≤11,-15≤l≤15范围内共收集到4037个衍射点,其中独立衍射点1442个(Rint=0.0251),1192个(I>2σ(I) )衍射点用于结构测定和修正。晶体结构由程序SHELXS-97和SHELXL-97直接法解出[21-22],经多轮Fourier合成获得全部非氢原子。
2 结果与讨论
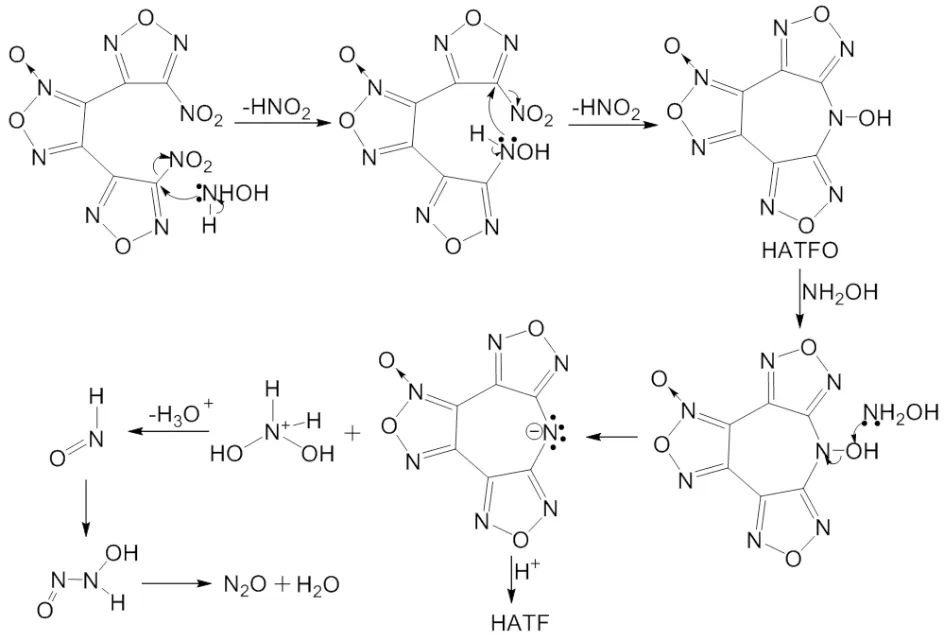
2.1 环化反应机理推测
呋咱环和硝基都有强吸电子性,导致与硝基相连的碳原子电子云密度降低,显示出部分正电性,有机碱NH2OH中孤对电子进攻C—NO2中显正电性的C原子,失去二分子HNO2,得到中间体7-羟基双呋咱并[3,4-b∶3′,4′-f]氧化呋咱并[3″,4″-d]氮杂环庚三烯(HATFO)。由于呋咱强吸电子性和七元环大共轭体系影响,导致N—OH电子密度更多流向氮原子,羟基氧显示部分正电荷。NH2OH中孤对电子进攻羟基氧原子,生成七元环阴离子和二羟基铵盐。七元环阴离子与质子结合生成目标化合物HATF,二羟基铵盐经脱质子和二次脱水后,最终分解成N2O,机理推测过程如下:
2.2 13C NMR和15N NMR核磁谱分析
HATF分子结构由于氧化呋咱环外配位氧的影响而不对称,配位氧将对整个分子碳谱和氮谱产生直接影响。目标分子在二甲基亚砜(DMSO)中具有较好的溶解性,获得了HATF在氘代二甲基亚砜溶液中的13C NMR和15N NMR谱图,如图1所示。
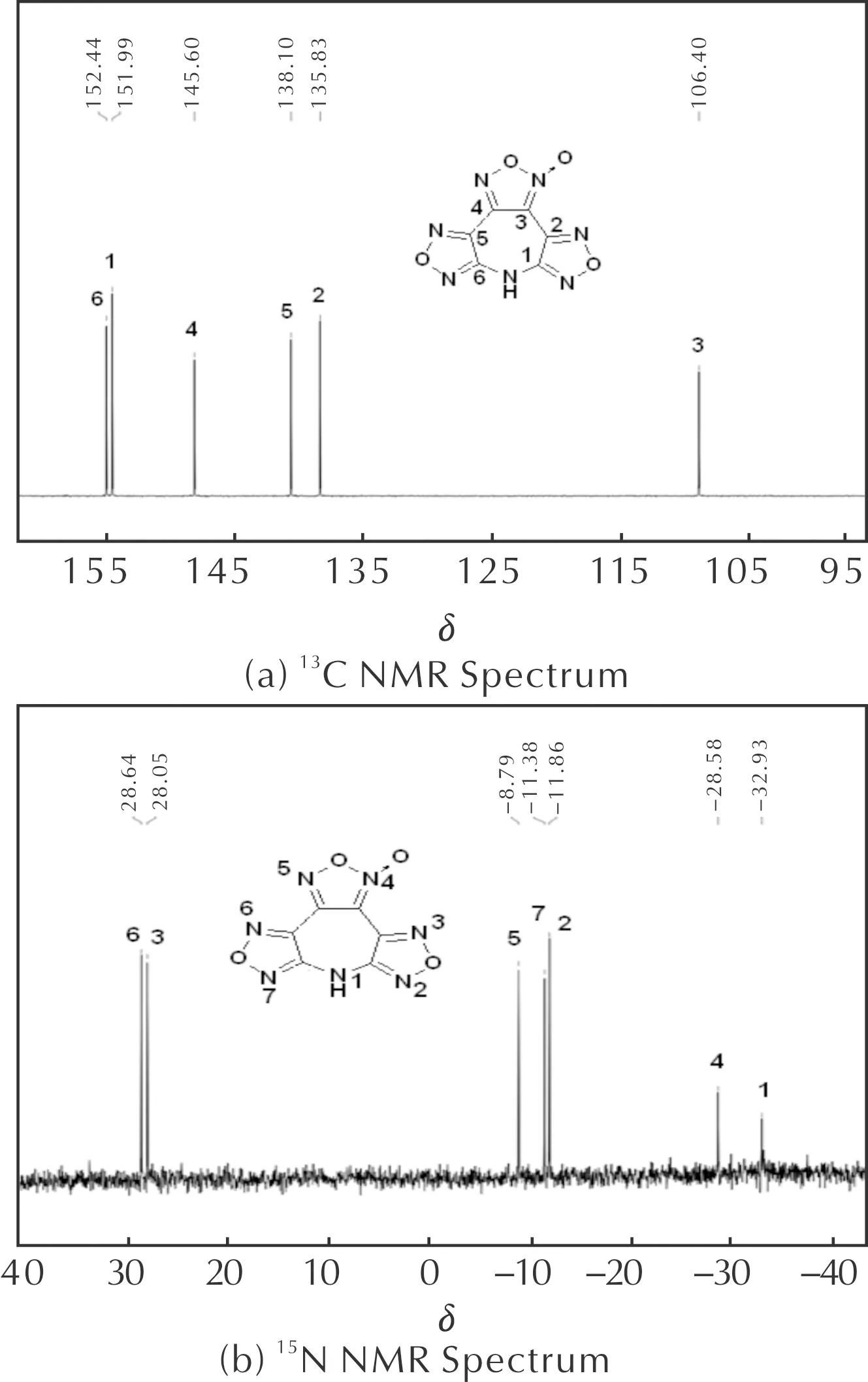
图1 HATF在氘代二甲基亚砜溶液中13C NMR谱和15N NMR谱Fig.1 13C NMR and 15N NMR spectra of HATF in DMSO-d6
首先,氧化呋咱环外配位氧对氧化呋咱上两个碳化学位移具有显著影响,基于大量文献数据可知,与环外配位氧接近的碳原子化学位移通常在δ为100附近,另外一个碳化学位移通常在δ为145左右,基于此,很容易将化学位移δ为106.40和145.60信号峰归属为C(3)和C(4)。由于配位氧与呋咱位置较远,电子效应影响较小且影响效应不明晰,因此仅仅根据文献很难对呋咱上碳谱进行准确归属,尤其是C(1)和C(6)、C(2)和C(5)的指认。规范不变原子轨道(Gauge Independent Atomic Orbital,GIAO)[23]方法是目前公认的预测核磁共振化学位移较为准确的方法,王民昌等[24]已成功用于一些呋咱含能分子的NMR预测。在结构优化基础上(B3LYP方法6-311+G(d, p)基组水平),用GIAO方法,对HATF分子中C、N原子的化学位移进行理论计算,结果得C(3)、C(2)、C(5)、C(4)、C(1)、C(6)的δ分别为108.4、138.7、141.4、149.4、155.5、156.5。通过与实验数据对照研究,实现了HATF分子13C NMR的归属(图1(a)),其中C(3)和C(4)理论值和实验值基本吻合,验证了方法的准确性。同理,完成了HATF氮谱归属(图1(b))。
2.3 晶体结构分析
HATF的晶体结构数据和精修结果见表1;部分键长、键角见表2。

表1 HATF的晶体结构数据和精修参数
图2和图3为HATF的分子结构图和晶胞堆积图。从图2可以看出,HATF为七元环状化合物,呋咱环、氧化呋咱环和NH形成了一个近似平面结构的分子。HATF平面分子之间距离d为3.20Å,分子间强烈的π-π作用力,驱使分子形成层状一维链,如图3(a)所示。另外,每个HATF分子上N(7)和H(1)分别与另外两分子的HATF形成两个N—H…N氢键,平均距离为3.01Å,夹角为167.1°;同时,每个HATF分子上O(2)和N(3)分别与另外两分子N(3)和O(2)原子之间存在范德华作用力,O…N之间距离为2.96Å,表明作用力较强。因此,晶胞中每个HATF分子分别与4个临近的HATF分子形成2个氢键和2个范德华作用,进而形成二维网状结构,如图3(b)所示。在分子间π-π作用、氢键及范德华力共同作用下,驱使HATF形成致密的三维堆积(图3(c)),揭示了HATF高晶体密度的内在原因(ρ=1.92g/cm3)。

表2 HATF的键长和键角

图2 HATF的分子结构Fig. 2 Crystal structure of HATF

图3 HATF的晶胞堆积图(a)分子间π-π堆积作用; (b)分子间氢键和范德华作用; (c)晶胞堆积图Fig.3 Crystal structure of HATF (a)π-π Interactions within HATF; (b) Hydrogen-bond and van der Waals′ interactions in HATF molecules; (c) Molecular packing of HATF molecules
2.4 热性能分析
采用差示扫描量热仪,在10℃/min的升温速率条件下研究了HATF在30~450℃条件下的热稳定性,结果如图4所示。

图4 HATF的DSC曲线Fig. 4 DSC curve of HATF
从图4可以看出,在178~187℃范围内,有一个峰型较钝的吸热峰,峰值为183.0℃,为HATF的转晶吸热峰;随后,在225~231℃范围内,有一个峰型尖锐的吸热峰,峰值为227.7℃,为融化吸热峰。随着温度继续升高,从271.5℃开始,出现一个缓慢放热峰,说明HATF开始分解,放热峰温为323℃,分解起始温度超过了270℃,由此表明该化合物具有优异的热稳定性,也侧面印证了平面环状化合物具有良好的热稳定特性。
2.5 爆轰性能分析
基于HATF晶体的结构构型,采用密度泛函理论(DFT)的B3LYP方法[26],在6-31+G(d, p)基组水平上对HATF的结构进行了全优化,经振动频率分析发现无虚频,表明优化结构为势能面上的极小点,为稳定构型。使用原子化方案[27],利用完全基组方法(CBS-4M)[28]计算了298K时HATF的焓H0(Molecule,298K),进而求得气相生成焓为776.6kJ/mol。利用静电势参数和Politzer等[29]提出的公式计算了HATF的升华焓ΔHsub(122.5kJ/mol),进而求得固相生成焓ΔfH(s, M, 298K)为654.1kJ/mol。
基于HATF晶体密度(1.92 g/cm3)和固相标准生成焓,采用EXPLO 5(V6.04)程序预估了其爆压为31.4GPa,爆速为8541m/s,并与RDX和HMX的文献结果进行了对比,结果见表3。由表3可知,HATF的密度高于RDX和HMX;HATF热分解温度高于RDX,与HMX相当。由于该分子结构中无硝基,故HATF的氧平衡低于RDX和HMX,爆速和爆压低于HMX。

表3 HATF的爆轰性能参数
3 结 论
(1)以3,4-双(4′-硝基呋咱-3′-基)氧化呋咱和羟胺水溶液为原料,获得了7H-双呋咱并[3,4-b∶3′,4-f]氧化呋咱并[3″,4″-d]氮杂环庚三烯(HATF)的一种新合成方法,提出了相关反应机理。探讨了HATF的13C NMR谱和15N NMR谱,基于实验结果和理论模拟,完成了目标分子13C NMR谱和15N NMR谱的归属。
(2)首次培养了HATF单晶,采用X射线单晶衍射研究了HATF一维、二维和三维结构特征,揭示了平面分子化合物HATF高晶体密度(1.92g/cm3)内在原因,提供了一种提高含能化合物密度的途径。
(3)HATF的计算生成焓为654.1kJ/mol,晶体密度为1.92g/cm3,起始分解温度为271.5℃,预估的爆速为8541m/s,爆压为31.4GPa,表明HATF是一种具有密度大、分解温度高、爆轰性能优异的高能量密度化合物。
猜你喜欢
爆破器材(2024年2期)2024-06-12 01:26:20
兵器装备工程学报(2022年12期)2023-01-06 04:24:04
化工管理(2021年7期)2021-05-13 00:46:24
火工品(2019年3期)2019-09-02 05:48:22
火炸药学报(2018年1期)2018-04-19 02:42:45
中国农资(2016年1期)2016-12-01 05:21:23
化工进展(2015年3期)2015-11-11 09:08:25
应用化工(2014年7期)2014-08-09 09:20:28
火炸药学报(2014年5期)2014-03-20 13:17:52
火炸药学报(2014年5期)2014-03-20 13:17:50
