
若尔盖高原泥炭沼泽湿地土壤细菌群落空间分布及其驱动机制
2021-04-25 05:18:32王好才刘圣恩展鹏飞
生态学报 2021年7期
王好才,夏 敏,刘圣恩,王 燚,展鹏飞,王 行,*
1 西南林业大学国家高原湿地研究中心/湿地学院, 昆明 650224
2 中国科学院沈阳应用生态研究所, 沈阳 110016
3 云南农业大学农学与生物技术学院, 昆明 650201
4 福建师范大学地理科学学院, 福州 350007
湿地生态系统是地球上最重要的生态系统之一,在物质循环、能量流动、维持生态平衡等方面发挥着重要的生态价值和环境功能[1-3]。微生物是湿地生态系统的重要组成部分,是土壤有机质与土壤养分转化与循环的动力,开展湿地土壤微生物相关研究对于认识和了解湿地生态系统生境状况与演化具有重要意义[4-6]。我国的高原湿地主要分布在海拔3000m以上的高原区域,是一类特殊的湿地类型。受气候变化及人为活动干扰,高原湿地生物组成及区系特征对环境变化异常敏感,其土壤微生物群落多样性和分布状况受土壤质地、植被类型、土壤pH及水分状况等因素影响[7]。因此,在全球气候变化背景下,开展高原湿地土壤微生物多样性及其空间分布研究,对进一步辨析湿地关键生态特征及其演化过程具有重要意义[8]。
土壤微生物具有明显的地域性特征,表现出一定的空间分布关系[9]。当前,土壤微生物群落结构相似性和地理空间距离的响应关系在草地、农田、森林等生境中已被证实[10-12]。在湿地生态系统中,有关土壤微生物空间分布特征的研究主要集中在人工湿地。如有研究表明,在受人类干扰较大的人工湿地类型(如库塘、水稻田等)中存在微生物群落相似性随地理距离增加而衰减的一般规律[13-14]。在自然湿地生态系统中,尤其是原始生境保存较好的高原泥炭沼泽湿地,土壤微生物群落相似性对地理距离的响应关系仍然未知。环境因素显著影响微生物群落组成和分布,微生物—环境之间存在密切的相互作用关系[15-17]。空间尺度大小对土壤微生物不同成员在群落构建方面的潜在驱动因素还尚不明确。在土壤微生物群落中,稀有种被定义为在不同样品间占据率较低的微生物,而丰富种是指在大量样品中普遍存在且丰度较高的类群[18]。在气候变化、群落组装过程及环境适应方面,稀有种和丰富种的响应机制表现出明显的差异[19-21],而在以往的微生物地理学研究中,往往会把所有微生物群落作为一个整体来探究其生物地理分布模式,少有将群落按照丰度高低划分为稀有种和丰富种,并分别加以研究。由此我们提出假设:在自然湿地生态系统中,微生物群落不同成员的生物地理分布模式可能存在明显的差异。
由于若尔盖高原泥炭沼泽湿地在碳存储方面的重要性,人们在全球变暖、水文波动环境下的土壤微生物活性及多样性变化方面开展了大量研究工作[22-23]。目前,对若尔盖高原湿地土壤微生物的研究侧重于生境退化和扰动对微生物多样性及结构的影响,以及泥炭层中有机物的微生物利用特征等方面,而土壤微生物生物地理分布模式尚不明确[24-26]。空间距离和环境异质性被认为是形成遗传变异和种群多样性的主要因素,然而在高原原生湿地土壤中,它们的相对重要性仍未得到充分认识。鉴于此,该研究以若尔盖高原泥炭沼泽湿地土壤细菌群落为研究对象,采用16S rDNA基因测序分析技术,探究高原泥炭沼泽湿地土壤细菌群落的生物地理分布模式,同时依据微生物群落在土壤样本中出现的丰度高低划分为稀有种和丰富种,研究群落内不同成员对地理分布模式的响应关系,以及影响群落多样性的环境驱动因素。该项研究工作的开展,有助于从微生物生物地理学的角度解释若尔盖高原泥炭沼泽湿地土壤细菌群落结构及多样性的分布规律和响应机制,为进一步研究若尔盖湿地土壤微生物地球化学循环过程,揭示微生物多样性对高原湿地生态系统功能的影响提供科学依据和理论基础。
1 材料与方法
1.1 研究地概况
若尔盖高原泥炭沼泽湿地位于青藏高原东北部,川甘两省交界处(33°25′—34°00′N, 102°29′—102°59′E),为黄河源区的重要组成部分,具有丰富的动植物资源和生物多样性。气候寒冷湿润,年均温在0.6—1.1℃之间,根据第二次全国湿地资源调查结果,研究区的湿地类型为草本沼泽,主要优势植物为木里苔草(Carexmuliensis)。若尔盖大沼泽原始生境保存较好,是我国第一大高原沼泽湿地,也是世界上面积最大、保存最完好的高原泥炭沼泽,是青藏高原高寒湿地生态系统的典型代表。作为重要的生态敏感区、高山生物多样性丰富带,若尔盖高原沼泽泥炭地在全球生态系统保护中具有重要地位,在全球碳储量、土壤水分涵养等方面发挥重要作用[27]。
1.2 实验设计与样品采集
由于若尔盖泥炭沼泽湿地呈斑块状分布,结合保护区湿地生境特征,于2018年8月挑选了原始生境保存较好的一块典型泥炭沼泽湿地作为研究对象,从局域尺度(local scale)开展微生物地理学研究工作。研究区沼泽水深在0.3—0.5m左右,为原生生境,在保护区内部不存在放牧干扰。深入原生沼泽湿地内部进行样品采集,结合前人采样策略[28]及前期研究基础[29],拟采用嵌套采样方案(图1)。分别设计3个100m2的样方,第1个样方与第2个样方之间的间隔为100m,第1个样方和第3个样方之间的间隔为1000m。每个100m2的样方内分别嵌套一个10m2和1m2两种尺度的小样方。在每个100m2的样方内,共取10个土壤芯(0—10cm),3个样方一共采集30个土壤芯(图1,见红色圆点所示)。该采样策略可以对土壤微生物β多样性的变化和更替进行有效评估[28]。在每个采样点设置0.5m×0.5m样方,将样方内部的植物地上部分收割后带回实验室,清洗干净并放入65℃烤箱中烘至恒重,用四分位电子天平进行称重得到地上生物量。随后利用荷兰Eijkelkamp定深泥炭钻采集0—10cm湿地表层土壤样品,使用环刀(7.5cm直径,5cm高)和自封袋分装。用镊子将环刀中的根系挑选出来,用淘洗法对地下根系冲洗干净后,放入65℃烤箱中烘至恒重,称重得到地下生物量。采集的土壤样品一部分放入冻存管后在便携式液氮罐中迅速冷冻,带回实验室后放入-80℃冰箱保存,用于土壤微生物分析。

图1 研究区域位置及采样点示意图
1.3 土壤理化性质的测定
土壤容重、土壤孔隙度、含水量等指标的测定使用环刀里的土样。泥炭土壤自然储水量以及最大储水量分别由自然含水率、饱和含水率换算得到。土壤总氮总磷采用浓硫酸消煮法,之后采用流动分析仪(AA3,Bran+LuebbeCrop,德国)测定;土壤硝态氮、土壤铵态氮采用氯化钾浸提法,之后采用流动分析仪测定;土壤pH采用玻璃电极法(MP511 pH计)按土水比1:2.5测定;土壤活性磷采用碳酸氢钠—钼锑抗比色法;土壤活性碳采用高锰酸钾氧化法;土壤阳离子交换量采用三氯化六氨合钴浸提—分光光度法。同时,使用TOC总有机碳分析仪(德国元素Vario)测定土壤溶解性有机碳;使用马弗炉仪器测定土壤灰分;用氢氟酸-高氯酸-硝酸消解法测定土壤K、Ca、Mg、S、Fe以及Mn 6种元素(微波消解仪,全谱直读电感耦合等离子体发射光谱仪(ICPE—9820));利用电感耦合等离子体发射光谱法同时测定土壤中可交换态钾、钙、镁。土壤基本理化指标测定结果见表1。

表1 研究区域土壤理化指标统计表
1.4 土壤DNA提取和16S rDNA扩增子测序
土壤微生物DNA提取使用Ezup柱式土壤基因组DNA抽提试剂盒,按试剂盒操作说明书提取,并采用0.8%琼脂糖凝胶电泳检测DNA。16S保守序列片段的扩增引物使用515F(5′—GTGCCAGCMGCCGCGGTAA—3′)和806R(5′—GGACTACHVGGGTWTCTAAT—3′)[30]。以10倍稀释后的基因组DNA为模板,根据测序区域的选择,使用带Barcode的特异性引物进行PCR扩增,每个样本进行三个PCR技术重复。PCR产物与1/6体积的6X loading buffer混合,使用2%琼脂糖凝胶进行电泳检测。对目的条带进行割胶回收,使用QIAquick Gel Extraction Kit(QIAGEN)试剂盒,回收后的PCR纯化产物使用Qubit@2.0 Fluorometer(Thermo Scientific)进行定量,最后等摩尔量混合。建库使用TruSeq DNA PCR—Free Sample Prep Kit试剂盒,构建好的文库经过定量和文库检测合格后,使用罗宁生物的Hiseq 2500平台PE250模式测序。
1.5 采样点的地理距离获取
每个采样点的空间地理坐标由手持GPS(eTrex enture, Garmin, Olathe, KS, USA)进行记录,之后采用平面直角坐标系来计算每个采样点的相对距离(以第一个采样点为原点,图1)。使用R语言中的Vegan包计算采样点之间的两两地理距离,从而创建采样点之间的地理距离矩阵。
1.6 数据分析与处理
1.6.1群落组成及Alpha多样性分析
使用R语言中的Vegan包进行数据预处理与alpha多样性和相对丰度的计算,将在所有样品中出现且丰度大于2的OTU定义为核心微生物(core microbiome),使用grid包和ggplot2包将数据进行可视化处理。
1.6.2土壤微生物群落相似性与地理距离的相关性分析
通过将全部OTU分类单元重新划分为稀有种(Endemic taxa)(在所有样品中出现的频率低于25%的OTU)和丰富种(Ubiquitous taxa)(在所有样品中出现的频率高于75%的OTU),分别产生4057个和716个OTU分类单元。随后,通过非度量多维标度(Bray—Curtis)距离[31],采用非参数多变量统计检验和相似性分析(999个排列),测定细菌总群落、稀有种群落、丰富种群落在局域空间尺度上不同样本之间的群落结构相似性[32]。细菌群落的周转速率用衰减率表示,该指标为地理距离(ln对数转换)与群落相似性(ln对数转换)关系的普通最小二乘回归(Ordinary Least Squares Regression)的斜率。斜率的计算公式如下:
ln(S)= ln(a)+zln(G)
式中,S为微生物群落相似性,G为地理距离,a为截距参数,z为衰减曲线的斜率,反映细菌群落的周转速率[33]。
1.6.3Mantel检验、方差分解分析
为了分析环境变量以及地理距离对微生物群落组成的影响,使用R语言中的ggcor包进行Mantel检验分析,检验不同群落组成和环境变量之间的Spearman相关性。同时,使用vegan包进行基于距离的冗余分析(Distance—based redundancy analysis,db—RDA)。
邻体矩阵主坐标(principal coordinates of neighbor matrices,PCNM)分析能够获得样本间空间关系的分解向量,显著的PCNM变量能直接解释空间尺度对物种的贡献[34]。PCNM分析通过对研究尺度范围内的空间距离建立截尾矩阵,通过主轴分析法进行处理,得到PCNM1、…、PCNMn,PCNM1表示整个研究尺度内的空间信息,属大尺度;PCNM2、…、PCNMn代表的尺度信息依次减少。得到空间因子数据后,结合土壤理化数据,采用方差分解分析(Variance Partitioning Analysis,VPA)来评估空间因子、土壤因子对细菌总群落、稀有种群落、丰富种群落多样性组成和结构变化的相对贡献。PCNM分析以及VPA分析均使用R语言中的vegan包进行。
2 结果与分析
2.1 土壤细菌群落丰度及多样性分析
2.1.1群落组成
在所有的土壤样本中,一共获得986532条高质量的细菌序列,在97%的相似水平下进行OTU聚类分析后,共得到6456个分类单元。通过将全部OTU分类单元重新划分为稀有种和丰富种,分别产生4057和716个OTU。我们比较了稀有种、丰富种和总细菌群落在门水平和属水平上相对丰度在前10的物种组成特征(图2)。
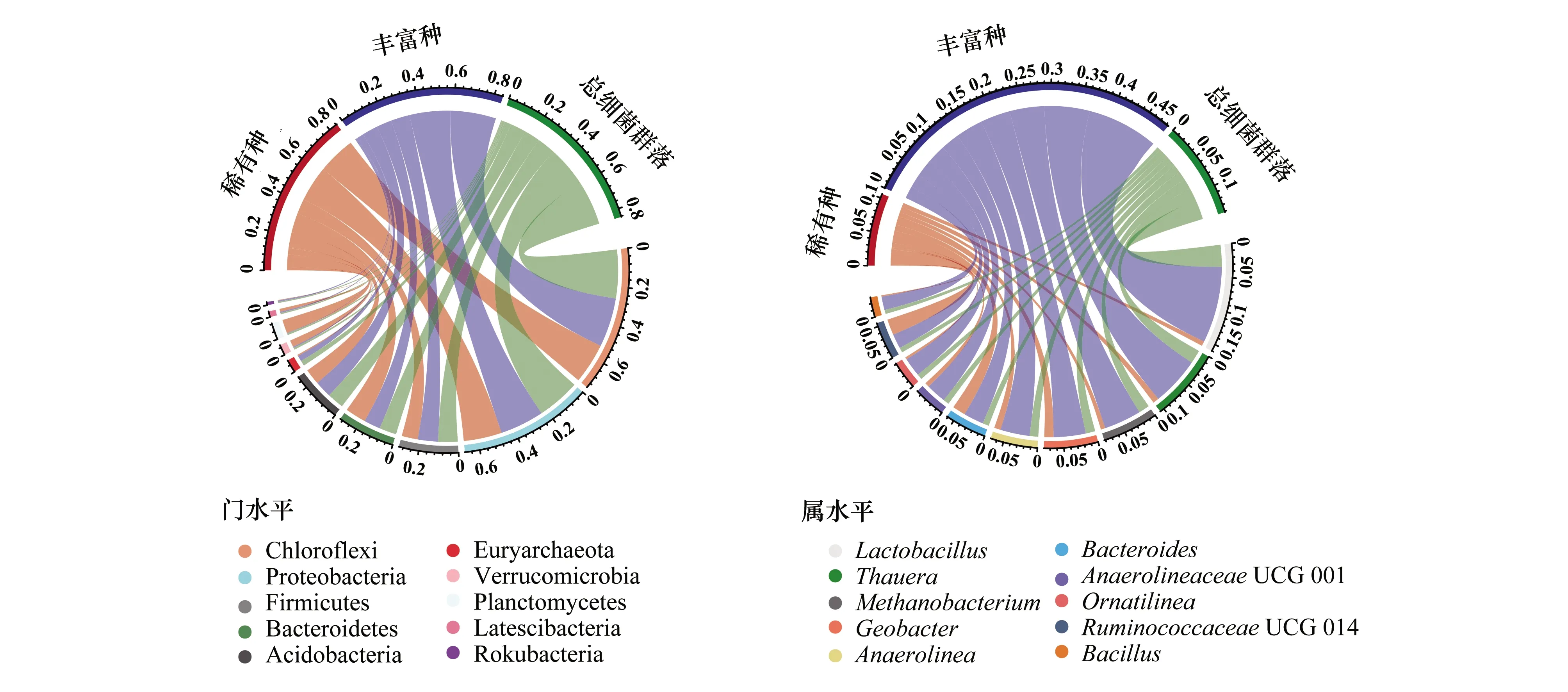
图2 稀有种、丰富种和总细菌群落在门水平和属水平上的相对丰度(前10)图
经微生物16S rDNA基因测序,共检测到54个菌门和934个菌属。其中,19个菌门和121个菌属的平均丰度高于1%。绿弯菌门(Chloroflexi)是显著的优势菌门,在稀有种群、丰富种群和总细菌群落中其所占丰度分别达21.88%、26.45%和26.25%,其他优势菌落分别为变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、酸杆菌门(Acidobacteria)、广古菌门(Euryarchaeota)、疣微菌门(Verrucomicrobia)、浮霉菌门(Planctomycetes)、Latescibacteria和Rokubacteria。在属水平上,乳杆菌属(Lactobacillus)是明显的优势菌属,在稀有种群、丰富种群和总细菌群落中其所占丰度分别达0.7%、11%和3%。在稀有种中,RuminococcaceaeUCG—014是优势菌属。
此外,通过30个土壤微生物样本信息,构建了基于泥炭土壤微生物的核心微生物群(core microbiome),共计278个OTU分类单元,分属于17个门水平和84个属水平。图3列举了核心微生物在门水平和属水平划分上相对丰度前10的核心微生物。其中,在门水平上丰度前三的细菌分别是绿弯菌门(Chloroflexi)、变形菌门(Proteobacteria)和厚壁菌门(Firmicutes),这些微生物在核心微生物群中所占丰度高达69%;在属水平上,丰度排前三的微生物分别是拟杆菌属(Bacteroides)、球菌科UCG.014(RuminococcaceaeUCG—014)和甲烷杆菌属(Methanobacterium),其所占丰度为18.04%。
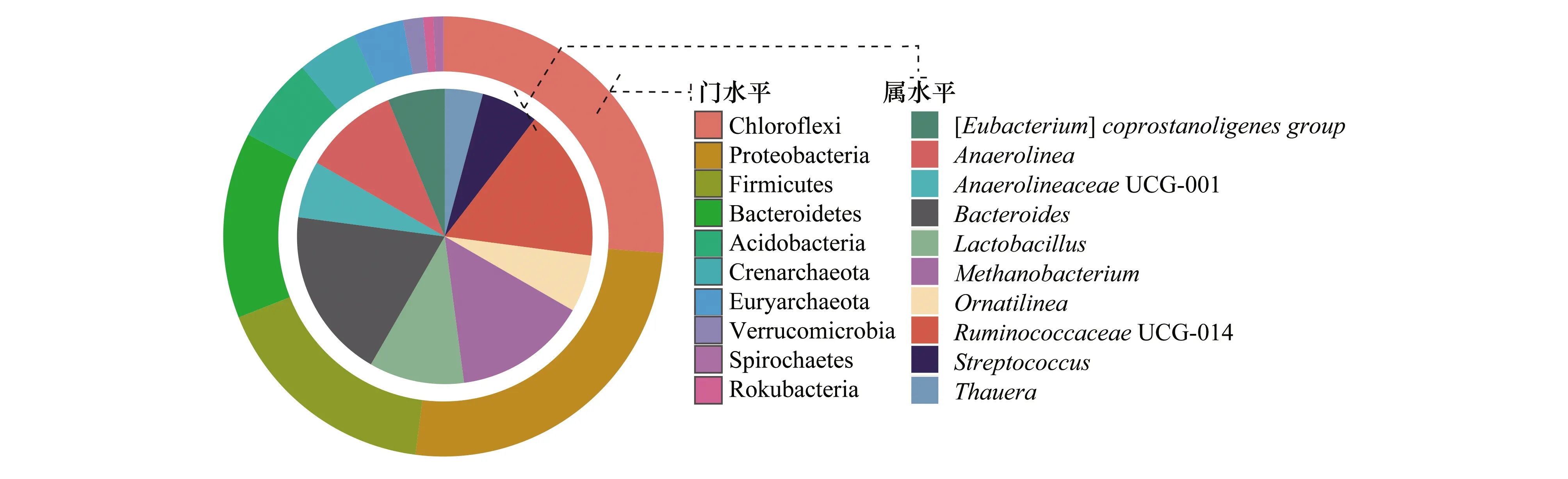
图3 基于30个土壤样本构建的门水平和属水平上相对丰度前10的核心微生物
2.1.2Alpha多样性
对研究区域所有采样点土壤细菌群落的Alpha多样性指数分析显示(图4),Chao1指数、Shannon指数和Simpson指数分别介于1663—3571、3.21—6.49和0.98—0.99之间,若尔盖高原湿地土壤样品具有较高的微生物多样性。同时发现,Chao1指数的变异系数(14.71%)大于Shannon指数(3.21%)和Simpson指数(0.32%)。
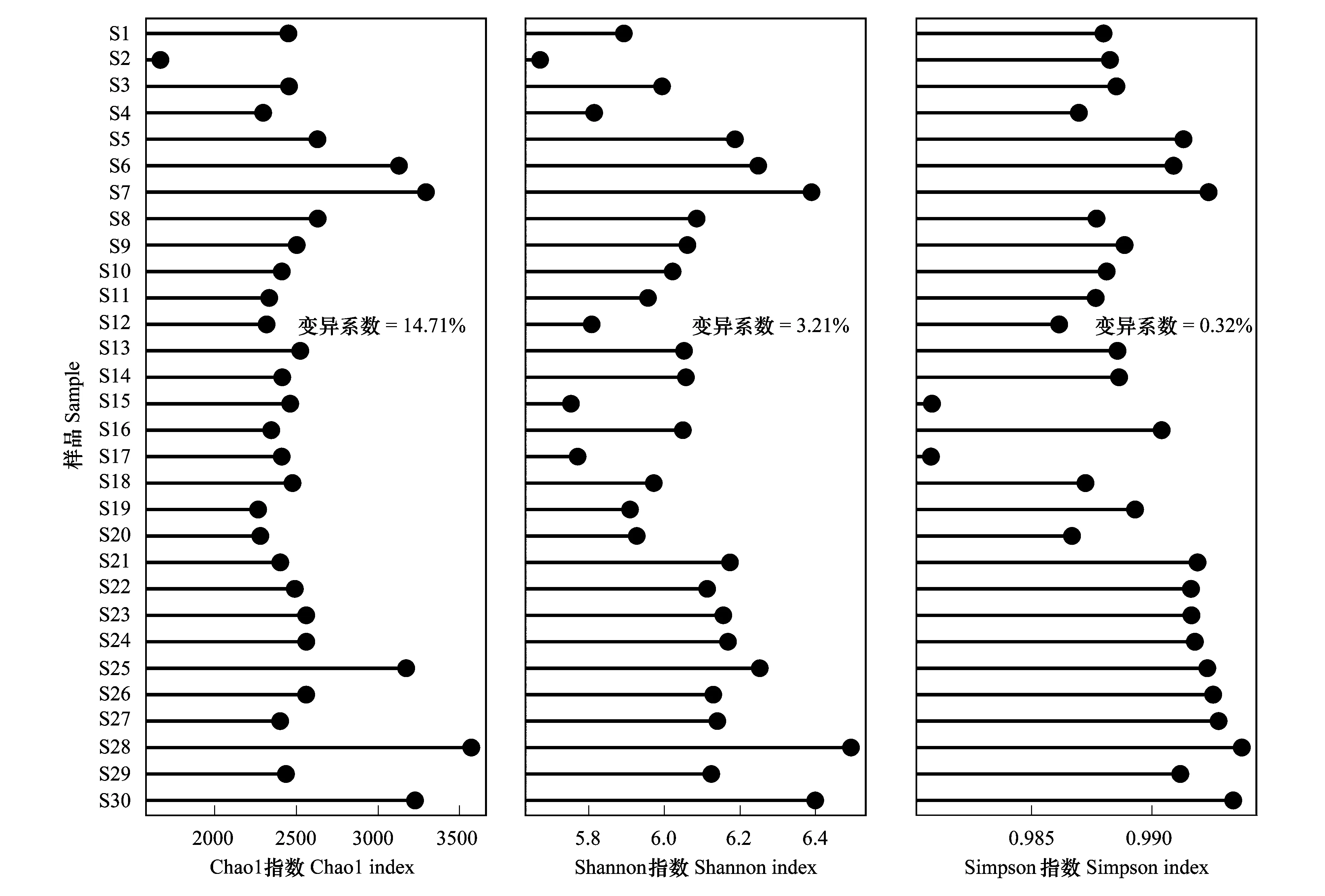
图4 细菌群落Alpha多样性指数变化Cleveland点图
2.2 细菌群落相似性的距离衰减特征分析
为探索高原泥炭原生沼泽湿地土壤中的微生物群落是否存在生物地理分布模式,对微生物群落相似性和地理距离之间的关系进行分析,并获得群落相似性随地理距离变化的衰减率特征。结果表明,稀有种、丰富种和总细菌群落的相似性均随着地理距离的增加而衰减(图5),表明高原泥炭湿地土壤微生物的空间分布特征符合生物地理分布的一般模式。通过比较,发现细菌群落的周转速率(图5,斜率Slope)在不同群落中存在差异。总体上,总细菌群落的距离衰减斜率(Slope=-0.026;P<0.001;R2=0.139)高于稀有种(Slope=-0.006;P=0.004;R2=0.056)和丰富种(Slope=-0.025;P<0.001;R2=0.112)。3个细菌群落对空间尺度的依赖性由大到小分别是总细菌群落、丰富种和稀有种。由此我们可以得出,在原始生境保存较好的若尔盖泥炭沼泽高原湿地生境中,土壤微生物群落的相似性随地理距离的增加而衰减,稀有种的周转速率则低于丰富种。
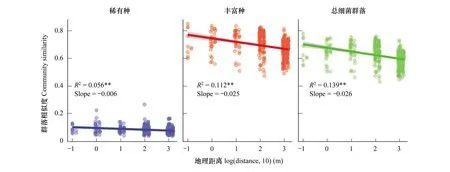
图5 群落相似性的地理距离衰减图
2.3 稀有种、丰富种和总细菌群落对环境因子的响应
对3种细菌群落和环境因子进行Mantel检验分析。结果显示(图6),地上生物量与3种微生物群落之间表现出极显著相关性(0.001 图6 环境因子和3类细菌群落的Mantel检验 为了进一步明确驱动微生物群落多样性在局域尺度上造成差异的关键环境因子,基于Bray—Curtis距离的约束主坐标分析表明(图7),稀有种、丰富种和总细菌群落多样性变异的11.54%、38.20%和31.85%可以用前两个主成分(CAP1和CAP2)来解释。按环境因子对3种细菌群落组成差异性影响的递减顺序(前向选择),这些显著因子分别为地上生物量、活性磷(丰富种、总细菌群落)和硫含量(稀有种),地上生物量是影响3种细菌群落组成多样性的关键环境因子;在稀有种群落中,CAP1与地上生物量呈显著负相关(R2=0.37,P<0.001),而在丰富种群落(R2=0.38,P<0.001)和总细菌群落(R2=0.43,P<0.001)中则为显著正相关。该分析结果进一步验证了Mantel分析检验结果。 图7 3种细菌群落和环境变量的冗余分析(db—RDA) 采用VPA方法分析环境因子(土壤环境因子)和空间因子(PCNM)对稀有种群、丰富种群和整体细菌群落组装的相对贡献。图8显示在3个细菌群落中,环境因子和空间因子对群落结构贡献的总解释量分别为4.93%、27.84%和26.63%,其中,空间因子在3种细菌群落的单独解释量分别为1.39%、3.59%和3.85%;环境因子在3个群落中的单独解释量分别为1.57%、7.27%和6.98%;空间因子、环境因子在3个细菌群落中的共同解释量分别为1.96%、16.98%和15.80%。总体上,在局域尺度上,环境因子对3种细菌群落组成差异性的贡献度要高于空间因子;稀有种群的VPA分析结果解释度明显低于整体细菌群落和丰富种群,可能是因为稀有种群落的产生具有较大的随机性,主要由随机作用(非决定作用)调控其群落构建(图5)。 通过对解释变量做前向选择分析(表2),发现空间因子PCNM1、PCNM2为主要解释变量(P<0.05),说明覆盖整个采样点的空间特征对样点之间微生物群落结构的差异具有显著影响;地上生物量和活性磷为丰富种和总细菌群落的主要解释因子,而在稀有种群中则为土壤硫含量和地上生物量。 前人针对若尔盖高原湿地土壤微生物开展了大量研究工作。如Zhong等[24]发现地下水位下降降低了原核生物群落的alpha多样性,微生物群落的垂向分布特征主要受地下水位波动影响。Tang等[6]研究表明在若尔盖地区,土壤类型对细菌群落的影响大于土壤深度对细菌群落的影响。Gu等[26]发现土壤退化导致若尔盖细菌和古菌群落的网络关联模式发生变化。Yang等[25]研究发现泥炭地退化减少了表层和深层土壤中的产甲烷菌的数量,甲烷排放下降。Cui等[22]研究表明温度增加了高寒湿地甲烷排放,而植被对甲烷菌群落组成有显著贡献,并与甲烷排放密切相关。目前,在若尔盖开展的研究工作主要集中在生境变化及扰动对微生物的影响,这些研究工作为我们进一步揭示若尔盖土壤微生物的地理分布格局打下了基础。 通过对比发现,不同区域的泥炭土壤细菌群落组成具有相似性。研究者[13]在对我国东北泥炭土壤细菌群落的研究中发现,酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、绿弯菌门(Chloroflexi)等为土壤中的优势细菌群落。而在本研究中,若尔盖泥炭土壤的优势细菌群落为绿弯菌门(Chloroflexi)、变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、酸杆菌门(Acidobacteria)等组成,在群落组成结构上与其他区域的泥炭土壤微生物组成具有相似性。基于前人的研究结果,我们认为若尔盖高原泥炭沼泽湿地中的这些土壤细菌群落是驱动若尔盖泥炭土壤生物地球化学循环的主要参与者,并同时参与了该区域湿地生境的形成与演化。然而,有关细菌群落在高原湿地生境中所发挥的具体功能仍需要进一步探索和研究。 虽然已有大量研究报道了不同生境中土壤微生物的生物地理模式,但少有研究关注人类活动干扰较少的高原湿地生境中的微生物生物地理模式,也少有研究阐明了微生物群落中不同类群群落相似度随地理距离的变化情况[10-13]。在本研究中,稀有种、丰富种和总细菌群落的相似度随地理距离的增加而衰减,证实了在高原泥炭沼泽湿地中土壤微生物也存在生物地理分布模式;3种细菌群落的距离衰减斜率在局域尺度上存在差异,且稀有种群落的相似度距离衰减斜率低于丰富种群,这与其他研究者在水稻土壤细菌群落中的发现有所不同[18]。在本研究中,稀有种群在30个土壤样品中的群落相似度低于20%,丰富种群的相似度则高于70%(图5)。稀有种在不同样品之间的差异主要由群落的演替、变更导致,而丰富种在不同样品之间的差异主要由群落的丰度高低导致。结合他们各自的距离衰减斜率规律(稀有种群落的相似度距离衰减斜率低于丰富种群),我们认为若尔盖高原泥炭土壤微生物相似性的地理衰减主要由高丰度的优势群落调控,物种丰度的变化是导致微生物群落地理距离衰减的主要因素。 环境因子是影响微生物群落结构的重要因素[11,17]。在本研究中,通过Mantel检验和冗余分析得出,地上生物量与3种微生物群落之间表现出极显著相关性(0.001 Jennifer等[33]通过比较盐沼沉积物中氨氧化细菌群落的组成,发现地理距离对局部尺度的微生物群落结构相似性影响显著,并认为距离效应的产生与生态漂移有关。然而,随着地理距离增加,微生物群落的地理衰减模式发生变化。Gao等[18]研究了水稻土细菌群落组装的空间尺度依赖性,发现稀有种群、丰富种群和总细菌群落在局域尺度(1—113m)、中观尺度(3.4—39km)和区域尺度(103—668km)内的空间依赖性存在差异。因此,采样尺度的大小是我们在研究微生物群落组成和微生物地理模式时不可忽略的重要因素[36]。在本研究中,我们在局域尺度上对不同细菌群落的空间尺度依赖性进行了研究,发现不同细菌群落的空间依赖性存在差异,从而进一步论证了前人的研究结果。此外,有研究者运用VPA分析揭示中国东部沿海地区水稻土壤和玉米土壤中影响古生菌群落构建的空间因素和环境因素,定量分析空间因子、土壤因子和气候因素对微生物群落构建的相对贡献[14]。研究发现,影响玉米和水稻土古生菌群落构建的相对贡献由大到小依次为土壤理化因子、空间因子和气候因子,该结果与本研究中得到的结果具有相似性(图8、表2),表明在我们所调查的局域尺度上,若尔盖泥炭湿地土壤微生物群落对环境异质性的响应较为强烈,是群落微生物构建的主要影响因素。 1)若尔盖高原泥炭沼泽湿地中土壤细菌多样性较高,共包括54个菌门和934个菌属。其中,19个菌门和121个菌属的平均丰度高于1%。在门水平上,绿弯菌门(Chloroflexi)、变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes)构成泥炭土壤核心微生物;在属水平上则为拟杆菌属(Bacteroides)、球菌科UCG.014(RuminococcaceaeUCG-014)和甲烷杆菌属(Methanobacterium)。 2)若尔盖高原泥炭沼泽湿地土壤细菌群落在局域尺度上存在较为明显的生物地理分布模式,即细菌群落相似性随地理距离的增加而衰减。在3种细菌群落中,群落的周转速率由大到小分别为总细菌群落、丰富种和稀有种。 3)地上生物量是影响高原泥炭沼泽湿地土壤细菌群落空间分布特征的关键环境因子;影响稀有种群落空间特征的环境因子还包括土壤硫含量、活性磷、Mn和土壤pH值。在局域尺度上,土壤理化因子对高原泥炭沼泽湿地土壤细菌群落构建的相对贡献大于空间因子。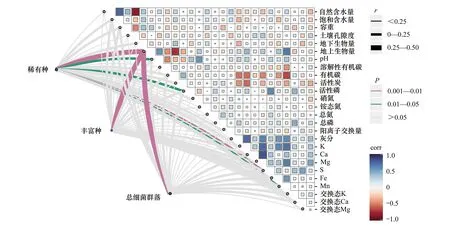

2.4 环境因子与空间因子对微生物群落构建的相对贡献
3 讨论
3.1 若尔盖高原湿地土壤微生物群落结构特征
3.2 若尔盖高原湿地土壤微生物地理模式
3.3 环境因子对土壤微生物群落的影响
3.4 环境因子和空间距离对土壤微生物群落构建的相对贡献
4 结论
猜你喜欢
成都信息工程大学学报(2021年1期)2021-07-22 07:21:50云南化工(2020年11期)2021-01-14 00:50:40家教世界·创新阅读(2020年4期)2020-06-03 04:38:56当代工人·精品C(2020年1期)2020-05-20 07:54:37家教世界·创新阅读(2020年1期)2020-05-11 11:47:03家教世界·创新阅读(2020年2期)2020-04-07 17:40:25中国化肥信息(2018年3期)2018-08-23 09:10:44青年文学家(2016年34期)2017-03-31 18:32:36中国诗歌(2016年7期)2016-12-05 20:43:22高原山地气象研究(2016年3期)2016-02-28 13:53:19
