
硅氧烷化合物的合成与应用进展
2021-04-21 02:14:28程玉桥赵文辉张治婷
材料工程 2021年4期
程玉桥,路 双,冯 喆,赵文辉,张治婷,赵 越
(天津工业大学 化学与化工学院,天津 300387)
随着材料科学的不断发展,硅氧烷化合物因其良好的耐热性、生物相容性、高透气性和高绝缘性能在诸多领域占据着重要地位。为了满足不同领域对功能材料性能提出的更高要求,不论在设计合成、反应机理以及物化性能等方面,基于硅氧烷化合物体系的研究日益受到科研工作者前所未有的关注。硅氧烷化合物通常是指含有重复硅氧键单元的化合物[1],由于硅氧化学键键能稳定、键长长、键角大、键易于旋转的特点以及硅氧烷与多种官能团构建分子的模式使得新型有机/无机硅氧功能材料发展为硅氧烷体系的重要部分。有机/无机硅氧功能材料主要包括线性结构(一维)、非线性结构(二维)、多面体低聚倍半硅氧烷(三维)以及有机/无机杂化硅氧烷化合物,传统的合成方法主要通过缩合聚合等方式制备硅氧功能材料,但易造成分子结构破裂且难以保证可控的分子量,近年来,科技工作者在优化改进传统方法和开发新方法方面开展了大量工作,得到了一定发展,但还不完善。因此,发展至今,精确控制硅氧烷化合物的分子量和结构依然是有机/无机硅氧功能材料的研究重点。本文在前期研究的基础上,系统地综述了硅氧烷化合物的研究进展,介绍了线性结构、非线性结构、多面体低聚倍半硅氧烷化合物以及有机/无机杂化硅氧烷化合物的设计合成方法,同时还讨论了它们在生物医学、航空航天、功能材料及三次采油的最新应用进展。
1 硅氧烷化合物的合成进展
根据硅氧烷化合物的分子结构可以分为以下几大类,主要包括线性硅氧烷化合物、非线性结构硅氧烷化合物、多面体低聚倍半硅氧烷化合物以及有机/无机杂化硅氧烷化合物。
1.1 线性硅氧烷化合物
线性硅氧烷化合物是基于硅氧烷主链的直链型高分子[2]。主链连接的有机取代基包括甲基、苯基、三氟丙基等惰性基团和氢、乙烯基或氨基等活性基团。惰性基团降低聚合物的表面能,活性基团有利于丰富硅氧烷化合物的多样性。其发展阶段包括均聚物、多嵌段共聚物以及可控分子量硅氧烷共聚物。传统的合成方法主要为环状硅氧烷单体开环聚合和线性含活性基团硅氧烷单体缩合聚合。考虑到此两种方法在合成精确可控聚合物方面的局限性,近年来均进行了不同程度的改进。Piers-Rubinsztajn反应是一种精确控制合成的特殊缩合聚合法。控制迭代法发展为合成线性硅氧烷化合物的新型方法。因此,目前线性硅氧烷化合物的设计合成方法包括开环聚合法、缩合聚合法以及控制迭代法等。
1.1.1 开环聚合法
开环聚合法是最普遍的合成线性硅氧烷化合物的方法。它是指由催化剂催化环硅氧烷单体开环聚合生成线性硅氧烷化合物。为便于构建新型硅氧烷结构,环硅氧烷单体逐渐发展成含功能基团的环单体衍生物。开环聚合包括阴离子开环聚合和阳离子开环聚合。传统的催化剂包括四甲基氢氧化铵和浓硫酸等。然而酸碱度过高易存在主链断裂、难剥离环状单体、催化剂难除等问题,不利于形成结构可控、分子量范围窄的线性硅氧烷化合物。近年来,有机酸碱催化剂被应用于精确控制合成硅氧烷化合物并且取得良好效果,特别是磺酸等有机酸和磷腈碱、胍类等有机碱。Shi等[3]以有机环三聚磷腈碱为催化剂,在室温条件下催化D4和八苯基环四硅氧烷(PDPS)开环共聚生成PDMS-ran-PDPS硅氧烷无规共聚物。经核磁表明高二苯基含量的硅氧烷化合物不含分支结构,因此有机碱磷腈碱催化有利于控制合成精确结构的硅氧烷化合物。Fuchise等[4]通过有机碱1,3-三亚甲基-2-丙基胍催化D3开环聚合,有机氯硅烷作封端剂合成窄分子量分布和所需末端结构的线性聚硅氧烷化合物。另外,此反应体系以水为引发剂,表明了非脱水溶剂也可用于合成结构明确的硅氧烷化合物。有机碱的催化活性取决于它们的Brønsted碱度和质子转移在链引发和链增长反应中的效率。有机环三聚磷腈碱的Brønsted碱度比胍类高,在水为引发剂的反应中,催化活性相对较弱的胍类催化剂发生副反应的概率低,更有利于合成结构可控的线性化合物。
1.1.2 缩合聚合法
传统的缩合聚合法主要包括氯硅烷水解缩合、同官能团缩合聚合以及异官能团缩合聚合。为了满足硅氧烷化合物精确合成的需求,Piers-Rubinsztajn反应(P-R)发展为缩合聚合法的特殊形式。P-R反应最初是指含氢硅氧烷化合物与烷氧基硅烷缩合形成新的硅氧烷化合物和副产物[5]。在催化剂三(五氟苯基)硼[B(C6F5)3]及不同反应活性亲核试剂作用下,可以在温和条件下得到高产率硅氧烷化合物。与易导致硅氧烷化合物结构破裂的酸碱催化剂不同,强路易斯酸B(C6F5)3优先与路易斯碱形成络合物,不稳定络合物和游离B(C6F5)3之间的微妙平衡会影响反应速率。随后,亲核试剂攻击络合物中的硅生成硅氧离子中间体[6],此中间体经分子内重排生成相应的化合物和副产物(烷烃或氢气)。除烷氧基硅烷之外,典型的亲核试剂发展为硅烷醇、芳基烷基醚和苯酚。水作为亲核试剂时反应速率相对较低。Madsen等[7]由3-氯丙基甲基二甲氧基硅烷和氢封端二甲基聚硅氧烷在B(C6F5)3催化条件下发生P-R反应生成共聚物,随后与乙烯基二甲基硅烷再次进行P-R反应合成具有乙烯基末端官能团的多官能团硅氧烷共聚物。此反应采用烷氧基硅烷作亲核试剂,生成副产物甲烷,当硅烷醇和酚作亲核试剂时则会生成氢气。Liao等[8]使用不同分子量的硅醇封端苯甲基硅氧烷与硅氢封端二甲基硅氧烷结合生成线性交替嵌段硅氧烷化合物。
1.1.3 控制迭代法
控制迭代法是精确合成低分子量线性硅氧烷化合物的新方式。它是指通过铱催化羰基化合物的氢硅化反应和P-R反应相结合快速多次迭代构建硅氧烷链。Matsumoto等[9]利用1,1,3,3,5,5-六甲基三硅氧烷与3-戊酮和二氢硅烷一锅法连续迭代18次反应后得到线性硅氧烷化合物(图1)。此方法通过顺序控制合成线性硅氧烷化合物,副产物影响较小。因为羰基化合物的氢硅化反应原则上不应形成任何副产物,P-R反应产生的副产物在这两种反应条件下通常都是惰性的。由于迭代次数最多仅为18次,故此方法只能获得低分子量线性硅氧烷化合物。
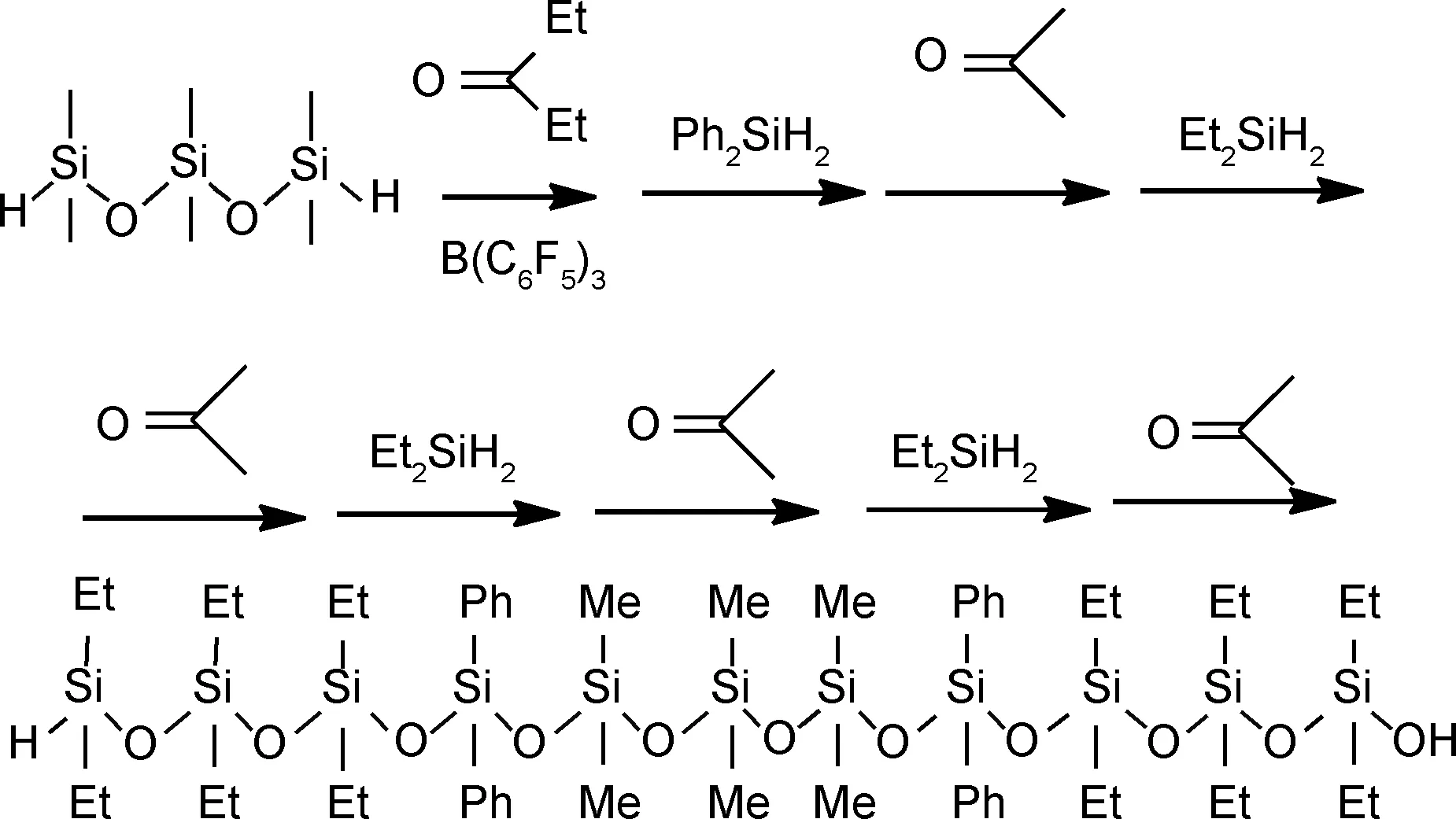
图1 控制迭代法合成线性硅氧烷化合物
1.2 非线性结构硅氧烷化合物
非线性硅氧烷化合物根据分子结构可分为以下几大类,主要包括环形硅氧烷化合物、超支化硅氧烷化合物以及含聚硅氧烷的互穿聚合物网络等。
1.2.1 环型硅氧烷化合物
环型硅氧烷化合物是指链端相互连接的环型拓扑结构化合物,其主要合成方法包括闭环聚合、扩环聚合以及P-R反应。闭环聚合是指由高稀释条件下分子链两端带有反应性官能团的低分子量硅氧烷化合物端对端环闭合,较高的稀释度有利于限制副反应发生程度。此方法很难选择性地合成单一的环硅氧烷化合物,需要经过复杂后处理分离出目标产物。扩环聚合是指将单体插入到现有的环状中间体的聚合反应。除以上两种传统方法外,Yu等[10]首次利用P-R反应合成了环状硅氧烷化合物。通过B(C6F5)3催化侧链含有Si—H键的二氢硅氧烷和三烷氧基硅烷化合物形成含烷氧基和硅氢功能基团的环四硅氧烷,随后进行分子间反应形成了环状硅氧烷化合物。此方法提供了将约束环结合形成环状化合物的新途径。此外,该聚合物可根据侧链取代基反应进一步构建新型复杂结构化合物。
1.2.2 超支化硅氧烷化合物
超支化硅氧烷化合物(hyperbranched siloxane compounds,HBS)由沿着聚合物主链的随机分散的树枝状单元、线性单元和末端单元组成[11]。基于高度分支拓扑结构和大量活性末端官能团的特点[12],功能化HBS逐渐被研究,目前合成方法主要为硅氢加成法、水解缩合法、酯交换法。
硅氢加成法主要以末端含有Si—H和烯烃基团的ABx型单体为原料在铂催化作用下合成产物,其中x确定每个重复单元的理想支链数。ABx型单体合成HBS易发生凝胶,难以控制聚合物的精确结构。多单体方法可以通过调整单体比例合成具有预期分子量的HBS。Wang等[13]采用了多单体四甲基二硅氧烷和乙烯基二烯丙基硅烷在Karstedt催化剂作用下得到超支化聚合物。此方法使用的过渡金属催化剂成本高,难以从终产物中去除。
水解缩合是设计合成超支化硅氧烷化合物的常用方法。此方法简单方便,单体来源广泛,逐渐应用于功能化超支化硅氧烷化合物。Liu等[14]先利用3-氨基丙基三乙氧基硅烷与3-缩水甘油氧基丙基三甲氧基硅烷反应。随后采用水解缩合法合成得到含羟基的HBS。
酯交换反应是指酯与醇在催化作用下交换生成新酯和新醇的反应,开始被应用于设计合成超支化硅氧烷领域。这种新合成方法经济高效且易于控制。Niu等[15]以四乙氧基硅烷、新戊二醇、三羟甲基丙烷和乙酰乙酸叔丁酯为原料通过两步酯交换反应生成含乙酰乙酰基的HBS。
1.2.3 含聚硅氧烷的互穿聚合物网络
互穿聚合物网络(interpenetrating polymer networks,IPNs)是指由两种或两种以上非均相聚合物组分经拓扑键交联形成的纠缠网络结构[16]。链缠结和交联的双重作用可以防止相分离并形成独特的形态和物理性质。含聚硅氧烷的互穿聚合物网络的合成方法分为顺序IPNs和同时IPNs。顺序IPNs是将预先形成的网络溶胀成含有合成第二网络需要的单体进而生成终产物,同时IPNs指两个网络被两种不同的独立化学路线同时合成[17]。Sano等[18]成功合成了聚丙烯酰胺和硅氧烷化合物的同时IPNs(图2)。硅醇键在硅烷化试剂条件下形成硅氧烷化合物,丙烯酰胺在交联剂作用下自由基聚合形成网络结构,这两种缩合反应同时独立进行,得到由聚丙烯酰胺凝胶和硅氧烷化合物组成的全IPNs型结构。
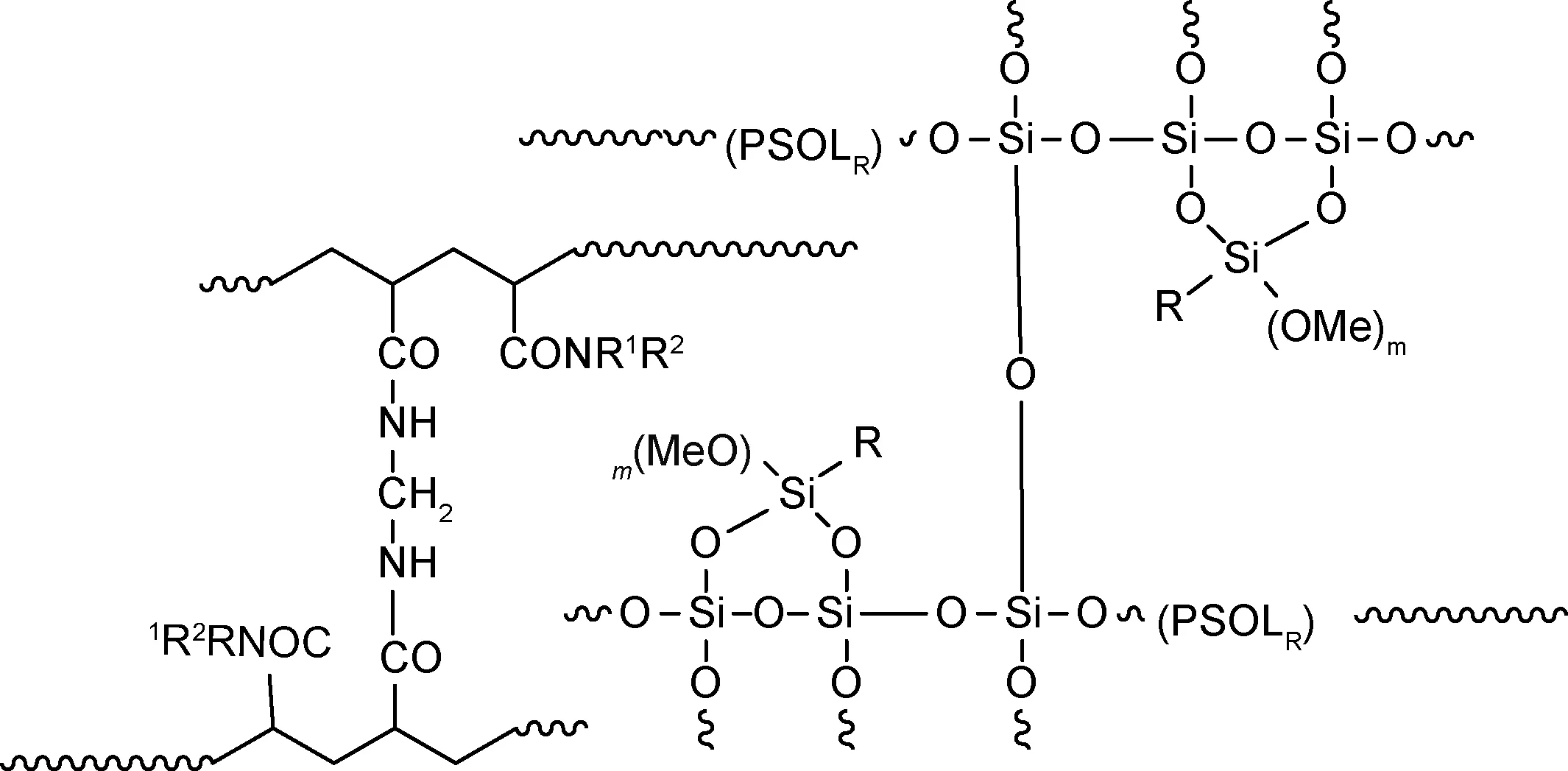
图2 同时IPNs法合成聚丙烯酰胺和硅氧烷化合物的互穿聚合物网络
1.3 多面体低聚倍半硅氧烷化合物
多面体低聚倍半硅氧烷(polyhedral oligomeric silsesquioxanes,POSS)是具有RSiO3/2重复单元的直径为1~3 nm的纳米结构化合物[19]。多面体的无机核可以为无规、梯状、笼状或部分笼状结构,如图3所示[20]。近年来,POSS化合物主要从无机核和笼外取代基两方面发展。双层倍半硅氧烷和金属多面体倍半硅氧烷结构丰富了无机核笼型、部分笼型和梯型的骨架体系。合成方法也在传统方法的基础上不断改进。POSS的合成方法主要为水解缩合法、顶点-盖帽法以及官能团衍生法[21]。

图3 多面体低聚倍半硅氧烷的结构分类
1.3.1 水解缩合法
以三烷氧基硅烷或三氯硅烷(RSiX3前体)为原料直接水解缩合是设计合成多面体低聚倍半硅氧烷(RSiO3/2)n衍生物最常用的方法。此方法简单高效、易于操作,但催化剂、反应物浓度和溶剂等因素影响较大。Wu等[22]以盐酸为催化剂将乙烯基三甲氧基硅烷水解缩合生成八乙烯基POSS。由于此方法水解速率低、缩合率高,在强催化剂存在下易得到完全缩合产物。而Choi等[23]利用碳酸钾催化剂通过控制苯基三甲氧基硅烷的不同初始浓度进行水解缩合,分别得到苯基取代的十二元笼状结构多面体低聚倍半硅氧烷和梯状聚苯基倍半硅氧烷。相比于LiOH和KOH等常见的强碱性催化剂,K2CO3的碱性较弱,有利于控制缩合产物的结构。通过改变催化剂和反应物浓度精确合成不完全缩合POSS是一种新途径。
1.3.2 顶点-盖帽法
顶点-盖帽法是指以含有三分子硅醇的不完全缩合倍半硅氧烷与R′SiCl3反应获得各种单反应性官能团POSS的方法。不完全缩合R7Si7O9(OH)3分子通常来源于三烷氧基硅烷/三氯硅烷部分水解或者完全缩合分子经四乙基氢氧化铵水溶液催化角裂解。此方法产率高、副产物少并且有助于将过渡金属元素引入硅氧骨架形成金属POSS。Maegawa等[24]以三氯硅烷为硅化剂使三硅醇乙烯基六异丁基取代的POSS封笼形成对位取代双乙烯基六异丁基T8笼型单体。Wu等[25]则利用钛酸异丙酯进行盖帽反应,形成Ti-NH2-POSS。金属Ti嵌入硅质笼的顶点可以显著增强热稳定性和阻燃性,同时新型金属POSS衍生物丰富了硅氧烷化合物体系。Tian等[26]以二甲基氯硅烷为硅化剂使双层硅倍半硅氧烷的多功能硅烷醇衍生物生成不对称硅氧烷化合物。双层倍半硅氧烷是指在对角位置含有两个反应性端基的硅氧烷化合物,其自身优良的分子柔韧性有助于进一步提高分散性和整体性能。经顶角-盖帽反应的化合物中依旧具备Si—H和Si—OH键,可用于进一步结构修饰。
1.3.3 官能团衍生法
官能团衍生法是指具备活性取代基的POSS通过取代、氧化及硅氢加成等反应转变成其他取代基的方法。Walczak等[27]利用硅氢加成反应衍生不同活性官能团的POSS,在Karstedt催化剂和{Ir(cod)Cl}2催化剂条件下两阶段反应合成双官能团POSS化合物(图4)。所得的衍生物扩大了双官能团硅氧烷化合物的范畴。由于存在与硅氧烷笼对称相连的两个不同基团,因此此物质具备进一步合成超支化分子或大分子的潜力。
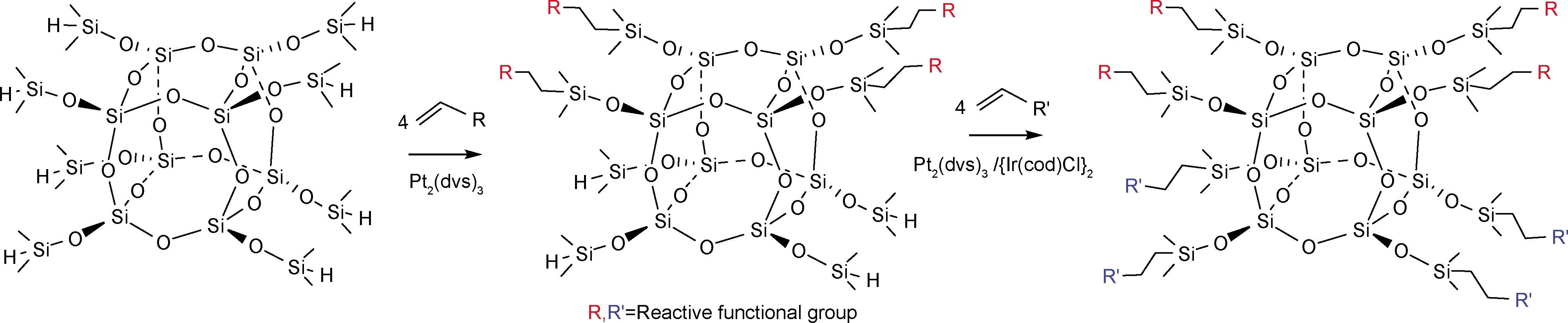
图4 官能团衍生法合成含活性基团多面体低聚倍半硅氧烷化合物
1.4 有机/无机杂化硅氧烷化合物
由于含有取代基的POSS作为无机构件结合在有机化合物结构上有利于实现纳米颗粒在聚合物表面或本体中的定向排列,因此发展有机/无机杂化硅氧烷化合物是极具吸引力的。根据POSS顶点上活性基团的数目,与有机化合物单体共聚可获得悬垂型、串珠型和星型有机/无机杂化硅氧烷化合物。发展至今,制备有机/无机杂化硅氧烷的方法有硅氢加成法、开环聚合法、原子转移自由基聚合法、可逆加成-碎裂链转移聚合法和点击化学法等。
1.4.1 硅氢加成法
硅氢加成法是指利用铂等过渡金属及其配合物为催化剂将硅氢键与不饱和键化合物加成结合形成硅氧烷化合物的方法,最常用的催化剂为Karstedt催化剂,是氯铂酸和乙烯基硅氧烷的络合物。该方法简单高效、活性高并且选择性好,但不利于控制催化过程,无法回收重复利用。Cao等[28]通过Karstedt催化剂催化八苯基二氢双层倍半硅氧烷和烯丙基缩水甘油醚反应得到串珠型双层倍半硅氧烷化合物。Cosgrove等[29]同样由Karstedt催化剂在二氯甲烷溶剂下催化聚二甲基硅氧烷和烯丙基七取代基POSS进行硅氢加成反应得到悬垂型有机/无机杂化硅氧烷化合物。
1.4.2 开环聚合法
开环聚合法在有机/无机杂化硅氧烷化合物合成中通常是指开环单体在含有羟基或氨基的POSS分子引发下生成目标产物的方法,开环单体通常为ε-己内酯和L-丙交酯。Qian等[30]由单羟基官能化的七乙烯基POSS引发开环单体ε-己内酯合成了以聚己内酯封端的七乙烯基POSS(图5)。Fan等[31]通过八羟基POSS衍生物引发L-丙交酯单体开环聚合生成星型有机/无机杂化硅氧烷化合物。
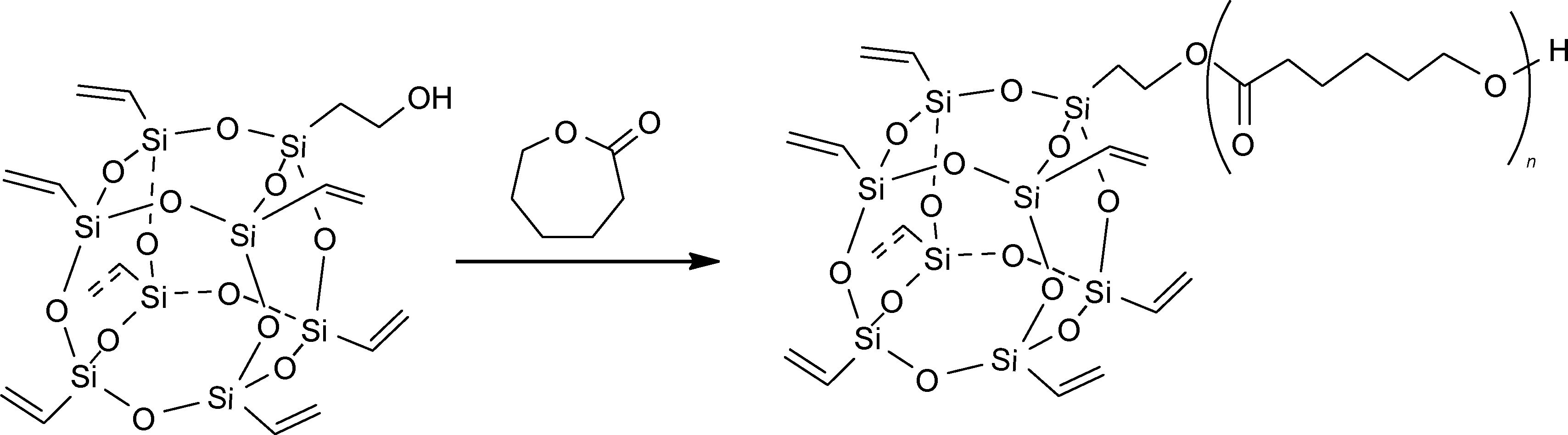
图5 开环聚合法合成有机/无机杂化硅氧烷化合物
1.4.3 原子转移自由基聚合法
原子转移自由基聚合方法是指利用铜(Ⅰ)卤化物催化剂反向活化休眠物种带有卤化物的RX(通常是烷基卤化物或卤化物封端的聚合物),促进烷基自由基的形成,引发乙烯基单体的聚合,从而形成碳碳键。此方法所需实验装置简单、催化剂价廉易得和单体丰富多样。缺点在于残留金属催化剂通常难以去除,限制在生物领域的应用。此反应合成有机/无机杂化硅氧烷化合物的引发剂主要包括卤化物封端大分子引发剂和卤化物封端POSS引发剂。Yang等[32]以单溴封端聚二甲基硅氧烷为大分子引发剂,引发甲基丙烯酸甲酯(MMA)和笼状甲基异丁酯POSS(MA-POSS),合成了PDMS-b-PMMAm-P(MA-POSS)n新型悬垂型结构杂化物。与大分子结构有限的卤化物封端不同,POSS封端卤化物作为引发剂可形成星型有机/无机杂化硅氧烷化合物。Hong等[33]以八(γ-氯丙基)-POSS为自由基引发剂,在氯化铜和五甲基二乙烯三胺催化体系下,和甲基丙烯酸三氟乙酯反应得到星型有机/无机硅氧烷化合物。
1.4.4 可逆加成-断裂链转移聚合法
可逆加成-断裂链转移聚合(reversible addition-fragmentation chain transfer,RAFT)法指直接使用加成碎片转移剂实现活性自由基聚合,此方法具备独特的无金属工艺、温和的反应条件和较高的转化率等优势。不同于常规自由基聚合反应,RAFT通过链转移剂控制聚合反应并且在聚合过程中链生长均匀可以很好地控制分子量分布。聚合机理的关键是加成-碎裂平衡。经引发剂引发的初级自由基向RAFT试剂链转移,经中间自由基断裂得到新的链转移剂和自由基,聚合结束后末端基团仍保持活性。有机/无机杂化硅氧烷化合物结构的成功合成很大程度上取决于RAFT链转移试剂的选择。典型的链转移试剂主要包括三硫代碳酸酯、二硫代酸酯和黄药化合物等物质。Li等[34]通过使用两端带有三硫代碳酸酯基团的聚二甲基硅氧烷长链为转移剂,与丙烯酸异丁酯POSS聚合反应合成了有机/无机杂化硅氧烷嵌段共聚物。Liu等[35]以3,13-二(2-溴丙酸)丙基双层倍半硅氧烷和1,4-二(黄药钾)丁烷反应生成的大分子链为链转移剂,随后与N-乙烯基吡咯烷酮发生可逆加成-断裂链转移反应获得化合物(图6)。三硫代碳酸酯类的链转移试剂适合聚合丙烯酸甲酯高活性单体,而N-乙烯基吡咯烷酮的活性较低,宜选用黄药类化合物。
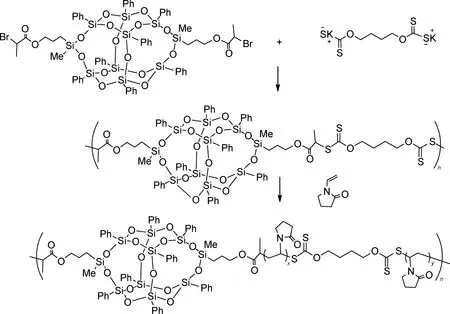
图6 黄药类化合物链转移试剂合成有机/无机杂化硅氧烷化合物
1.4.5 点击化学法
点击化学法是一种通过不同小单元结构分子模块化快速精确合成目标产物的方法,具备对水和氧气不敏感、产率高、原料来源广泛和副产物易被移除等特点。考虑到POSS分子在碱性或其他苛刻条件下不稳定,点击化学法是设计合成有机/无机杂化硅氧烷化合物而不损害POSS核心结构和功能完整性的理想工具。POSS作为纳米构件利用点击化学法的前提是需确保其含有可点击基团,如烯烃、环辛烯、炔类、叠氮化物、硫醇、胺类等。典型的“点击”反应包括Diels-Alder环加成、Cu(Ⅰ)催化的叠氮-炔环加成(CuAAC)和硫醇-烯反应等。Diels-Alder点击反应是指富电子双烯体和缺电子的亲双烯体之间直接环加成形成稳定的环己烯产物[36],此反应是热可逆的。Behera等[37]通过Diels-Alder点击反应将呋喃侧基官能化的聚氨酯(FPU)和马来酰亚胺官能化的多面体低聚倍半硅氧烷(POSS-M)反应生成FPU-POSS-M化合物。与CuAAC反应相比,使用高效Diels-Alder反应的优点在于不需要催化剂。Doganci等[38]以含叠氮基官能团的八臂聚(ε-己内酯)星型聚合物和炔烃末端官能化聚甲基丙烯酸甲酯为原料,进行CuAAC点击反应时则需要在CuBr/五甲基二亚乙基三胺催化体系下生成AB型杂臂星型聚合物(图7)。Zhang等[39]以八乙烯基POSS和3-(二甲基氨基)-1-丙硫醇(DPT)通过硫醇-烯点击反应生成POSS-(DPT)8,随后引入1,3-丙磺酸内酯进行季铵化亲核开环点击反应生成可溶性POSS八官能化衍生物。除硫醇-烯点击反应之外,硫醇基点击反应还包括硫醇-炔、硫醇-迈克尔和硫醇-环氧等反应。
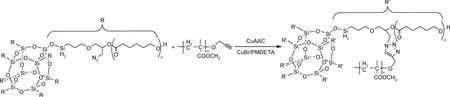
图7 叠氮-炔环加成法合成AB型杂臂星型有机/无机杂化硅氧烷化合物
2 硅氧烷化合物的应用进展
2.1 生物医学
由于聚硅氧烷弹性体具有良好的生物稳定性、低毒性和生物相容性,被普遍应用于生物医学领域,主要包括人造皮肤、义齿矫正器、柔性应变传感器[40]以及组织支架等[41]。人体皮肤是一种能够在创伤愈合后保持其力学性能和传感功能的软生物材料[42],因此,开发具有自愈能力的可拉伸聚硅氧烷弹性体是极具吸引力的。自愈现象的内在机理在于可逆共价键的形成,低玻璃化转变温度使弹性体具备理想灵活性,从而有利于提高链的流动性、形成足够浓度的可逆键以及保持弹性体的自愈性能[43]。硅氧烷化合物由于其较低的玻璃化转变温度、固有的高延展性和良好的热稳定性而被选为自愈弹性体聚合物的骨架。调整硅氧烷段的链长可以改变自愈弹性体的性能。具有容易调节应力松弛或蠕变依从性的硅氧烷基弹性体应用于生物医学形状记忆材料是研究的新方向。尽管硅氧烷化合物力学性能很差,但硅氧烷骨架的高键能与其惰性甲基的结合使硅氧烷化合物具有独特的生物稳定性和化学惰性,可与具备优越力学性能的物质共混,应用于皮肤组织工程支架,硅氧烷化合物的掺入也有利于提高共混支架的血液相容性[44]。
2.2 航空航天
严酷恶劣的太空环境和航天器超高速飞行过程产生的气动加热现象决定了普通材料难以应用于航空航天领域。硅氧烷化合物因其优良的耐高低温[45]、耐臭氧以及耐烧蚀性能被用于合成飞行器的防热材料和燃油系统的密封垫圈[46]。此化合物固有的弹性可以减轻剧烈发射振动的影响并保护隔热系统免于黏结。飞行器上升过程中的运载工具涂覆硅氧烷化合物涂层以抵抗气流与机身摩擦产生的超高热量。检查维修是保障航天飞机安全运行的必要环节,这无疑需要耗费很高的时间成本和经济成本。机身使用的复合材料在一定周期内会发生微裂纹,裂纹经生长合并后很有可能造成严重事故,硅氧烷化合物的自愈合性能和耐疲劳性有利于微裂纹传播前的自修复[47]。在复合材料中掺入低玻璃化转变温度的硅氧烷化合物以增强自愈合能力具备良好的应用前景。
2.3 功能材料
随着现代材料科学技术的不断发展,许多基于硅氧烷化合物的功能材料逐渐被研究。经荷叶效应的启发,超疏水超疏油材料引起了科技工作者的关注。这种材料不仅对水具有防潮能力,而且能保护表面不能被有机油污染[48]。由于荷叶表面大量的微纳米结构与液滴形成的空气缝隙是荷叶不能被液滴浸湿的原因,所以超双疏材料是低表面能和粗糙表面(微纳米结构)共同作用的结果[49]。硅氧烷化合物是合成超双疏功能材料常用的低能物质。材料表面独特微/纳米结构的硅氧烷化合物层是经硅烷水解缩合交联而制得的。具有可水解基团的硅烷与水反应形成硅烷醇,随后硅烷醇与底物面的羟基偶联,通过化学键的强界面作用可以将分子均匀且紧密地接枝在底物面上。剩余的硅烷醇能够进一步彼此缩合以生成具备独特微/纳米结构的硅氧烷化合物层[50]。另外,硅氧烷功能材料还具备防污能力。船舶的表面易受海底污垢的影响造成摩擦阻力和水动力阻力,从而增大燃料消耗成本和温室气体排放增加[51]。功能材料表面纳米结构基底和水滴之间的气穴使复合固体/空气/液体界面形成,液滴的接触角增加有利于表面的污垢被带走,此表面现象显著降低了污垢的附着力。
2.4 三次采油
尽管新能源迅速发展,石油在能源领域仍占据着重要地位[52]。然而,随着石油需求量日益增加,普通的表面活性剂驱油技术在复杂恶劣的油藏环境中难以保持良好的采油效果[53]。相较于普通表面活性剂在高温高盐储层中易被降解失活的特点[54],驱油用硅氧功能材料在具备优异耐温抗盐性的同时进一步作用于被束缚在油藏孔道中的残余油。此残余油主要受毛细管压力和油/岩吸附力的作用[55],驱油材料则需要解决水/油界面和油/岩界面两个方面的问题。一方面,驱油用硅氧功能材料具有较强的两相分布效率,接触到储层岩石孔隙中油滴时可显著降低油水界面张力,油滴通过狭窄孔喉时受到的毛细管压力随之降低。水包油乳液或者油包水乳液的形成有利于提高波及效率[56]。另一方面,驱油用硅氧功能材料的硅氧烷疏水链可以和岩石表面吸附的原油组分形成相互作用,甚至形成离子对,有效改善储层表面润湿性[57],进而降低石油在岩石表面上的吸附力,使油膜更容易被剥离且尽可能不产生二次吸附。故硅氧烷化合物可以应用于三次石油开采领域,大幅度提升石油开采量。
3 结束语
近年来,科技工作者致力于优化传统方法和开展新方法的研究,设计合成了结构多样的新型硅氧烷化合物,不断丰富和完善硅氧烷化合物体系,在控制合成有机/无机硅氧功能材料方面取得很大进展,拓宽了硅氧烷化合物的应用领域,但与当前经济社会发展需求仍有较大的距离,因此需要从以下三个方面继续深入研究:
(1)增强分子结构的精确控制与设计合成,构建结构迥异的具备精确分子量和结构的硅氧烷化合物目前还存在着一定难度。
(2)开发简单高效、成本低廉的合成方法,硅氧烷化合物的制备方法受诸多因素影响较为复杂,探索简单的合成路线间接降低了工业设备的成本,是实现产业化的必要途径。
(3)加强基础理论与产业化研究,进一步探索硅氧烷化合物的设计合成作用机理,加快产业化发展进程,以实现更多领域的应用。
猜你喜欢
陕西中医(2018年6期)2018-08-29 00:43:34
电子测试(2018年1期)2018-04-18 11:52:24
中国塑料(2016年1期)2016-05-17 06:13:00
读写算·教研版(2016年8期)2016-05-07 11:52:08
中国塑料(2016年11期)2016-04-16 05:25:55
合成化学(2015年4期)2016-01-17 09:01:04
中国塑料(2015年3期)2015-11-27 03:41:59
海军航空大学学报(2015年1期)2015-11-11 17:22:41
中国塑料(2015年10期)2015-10-14 01:13:16
中国塑料(2014年9期)2014-10-17 02:48:22
