
Class2 CRISPR-Cas 系统发掘及分析方法
2021-04-15 01:06朱晓菲黄娇媚
热带生物学报 2021年1期
朱晓菲,黄娇媚,原 昊,万 逸,3
(1.海南大学 海洋学院/南海海洋资源利用国家重点实验室,海口 570228; 2.海南大学 信息与通信工程学院,海口 570228; 3.中国科学院 海洋研究所/山东省腐蚀科学重点实验室,山东 青岛 266071)
Clustered Regularly Interspaced Short Palindromic Repeats-associated gene(CRISPR-Cas)全称为成簇的规律间隔的短回文重复序列,最初于1987 年在大肠杆菌中发现。ISHINO Y 等[1]在研究大肠杆菌iap(碱性磷酸酶)基因时,在其编码区3′端侧翼序列中发现长度为29 bp 高度保守的重复核苷酸序列,重复序列的间隔为32 bp。随着对该序列的深入研究,发现该重复序列广泛存在于古细菌和细菌的基因组中,直到2002 年JANSEN R 正式命名该重复序列为CRISPR 序列,除此之外,该研究还发现CRISPR基因的侧翼序列中有4 种同源基因(CRISPR-associated gene):cas1、cas2、cas3、cas4,这些基因编码一些功能蛋白,与CRISPR有功能相关性[2]。随着深入研究,CRISPR-Cas 系统的功能的免疫功能逐渐被发现,CRISPR-Cas系统类似于真核生物的RNA 干扰(RNAi)[3],后经证实是细菌对噬菌体等病原生物的获得性免疫作用[4]。CRISPR-Cas 系统在细菌对抗噬菌体侵入时分为3 个阶段。第1 阶段为适应阶段:在噬菌体侵入细菌时,Cas1-Cas2 蛋白复合物根据前间隔序列临近基序(PAM)位点将噬菌体靶DNA(protospacer)切割并将这段靶DNA 序列插入到CRISPR 重复序列5′端的末尾,产生新的间隔序列(spacer)。第2 阶段是基因的表达和处理阶段,间隔序列(spacers)和CRISPR 重复序列共同进行转录,形成初转录产物pre-CRISPR RNA(pre-crRNA),后由Cas 蛋白复合物对转录初产物进行切割,得到成熟的包含间隔序列(spacers)和重复序列的CRISPR RNAs(crRNAs)。不同的CRISPR-Cas 系统对pre-crRNA 的处理存在差异,有些由多个Cas 蛋白亚基处理,有的由单个Cas 蛋白处理,有的借助于宿主细胞的RNase。第3 阶段为干扰阶段,在guide RNA(crRNA 和tracrRNA 合成的引导RNA)的引导下,利用单独Cas 蛋白或是Cas 蛋白复合物对靶DNA 或RNA 进行切割。第一类CRISPR-Cas 系统在切割靶链时需要多个Cas 蛋白复合体的参与,而第二类CRISPR-Cas 系统在切割靶DNA 或RNA 时只需要单个Cas 蛋白加guide RNA(gRNA)即可完成对靶链的切割。因此,第二类CRISPR-Cas 系统成为现在基因编辑中重要的工具。
1 CRISPR-Cas 的发掘方法
Cas 蛋白作为CRISPR-Cas 系统中的切割靶链的效应部分,是寻找新CRISPR-Cas 系统的重点。目前,基于生物信息学手段发掘Cas 系统主要分为2 种方法,一种是基于对已知Cas序列建立隐马尔科夫模型(Hidden Markov Model,HMM)对细菌和古细菌基因组进行分析(图1a)。另一种是基于CRISPRCas 系统中的标志序列对细菌和古细菌的基因组进行Cas基因的查找(图1b)。

图1 基于生物信息学手段发掘Cas 系统的2 种方法Fig.1 Two methods to explore CRISPR-Cas system based on bioinformatics
对细菌和古细菌的CRISPR-Cas 系统发掘,所有的生物信息学分析都需要基于它们的基因组。CRISPR-Cas 系统广泛存在于细菌和古细菌中,在收集数据时需要分为两部分。一类是基因组数据库的收集,可以通过NCBI,EBI 等数据库进行细菌和古菌的全基因组数据收集和批量下载[5]。第二类是宏基因组,宏基因组由于数据库庞大,在Cas 酶发掘中收集方式多样,多数通过各种野外研究发现的数据进行基因组分析[6],宏基因组数据需要组装后才可以进行下一步分析。
1.1 建立隐马尔科夫模型进行CRSPR-Cas 的生物信息学发掘
1.1.1 读取基因组开放阅读框(Open Reading Frame, ORF)开放阅读框是指DNA 序列中具有编码蛋白质潜能的序列,从起始密码子开始,终止于终止密码子。通过读取开放阅读框,可以从细菌和组装好的古细菌基因组中识别出所有可以编码蛋白的潜在基因序列,目前应用于识别原核生物基因组开放阅读框频率较高的预测软件有Prodigal[7]、Glimmer[8]和GeneMark[9]等,软件优缺点对比见表1。其中,Prodigal 是在发掘新Cas 酶中明确提及使用过的开放阅读框识别软件[10]。准确的识别开放阅读框并对开放阅读框的位置进行准确定位有助于后续对CRISPR 序列定位后二者共同分析。

表1 ORF 预测软件对比Tab.1 ORF prediction software comparison
1.1.2 对已知的Cas 酶建立隐马尔科夫模型隐马尔科夫模型是一种统计分析模型,近年来被广泛应用到各种生物信息学分析中,主要用于描述某一核苷酸序列从其特定的祖代遗传而来的概率。根据现有的序列通过计算机对序列的分析建立隐马尔科夫模型,进而推测出最有可能出现的祖代序列[11]。
在用HMMER 软件建立隐马尔科夫模型之前,需要对准备建立模型的已知Cas 蛋白序列进行多序列比对。目前应用于多序列比对有以下几种软件,分别为CLUSTAL W、MUSCLE、T-COFFEE、DIALING2、MAFFT 等,软件速度MUSCLE 最快,对比准确性MUSCLE 最高[12]。通过对已知Cas 酶的多序列比对,得到STOCKHOLM(sto)文件,作为接下来的模型建立输入文件。
HMMer 是基于隐马尔科夫模型建立的生物信息学分析软件[13],有网页版和本地版,通过hmmbuild指令和Cas 蛋白多序列比对结果输入文件建立已知Cas 蛋白的隐马尔科夫模型,hmmsearch 指令和建立的Cas 蛋白模型输入文件可以对预测出的开放阅读框文件进行序列分析,进而推测出可能是Cas 蛋白的编码序列。
1.1.3 CRISPR 序列识别CRISPR 序列包含间隔序列和重复序列,是CRISPR-Cas 系统中另一个重要的组成部分。应用于发掘CRISPR-Cas 系统的目前有3 种,分别为CRISPRDetect[14]、CRISPR Finder[15]和PILER-CR[16]。其中,CRISPR Finder 应用最广[6,10,17],可以准确识别出长度短的CRISPR 序列,在升级后不止可以识别CRISPR 序列,还可以通过自带的隐马尔科夫模型对输入的序列进行Cas 蛋白的预测[18]。识别CRISPR 序列软件优缺点对比见表2。

表2 CRISPR 序列识别软件对比Tab.2 Comparison of CRISPR sequence recognition software
1.1.4 筛选在对基因组进行生物信息学分析后,得到软件预测出的Cas 蛋白和CRISPR 序列。对得到的候选序列进行筛选,筛选条件有以下3 条:1)同时含有Cas1 和CRISPR 序列;2)与Cas1 相邻的10 个ORF 之一包含1 个大于800 个氨基酸的未被鉴定的蛋白序列(通过隐马尔科夫模型预测出的);3)在同一基因组列中没有已经被鉴定出的包含Cas基因的干扰模块[10]。
1.2 以Cas1 和CRISPR 序列为标志序列进行CRISPR-Cas 系统的生物信息学发掘JANSE 等人的研究表明,有些CRISPR 序列上下游无编码Cas 的序列,有些编码Cas 酶的序列上下游无CRISPR 序列[2],因此,以Cas1 蛋白和CRISPR 序列为标志序列分别进行识别可以有效搜寻到所有候选序列。此种方法是根据已经发现的Class2 CRISPR-Cas 系统的结构特征进行发掘。
1.2.1 选取标志序列对数据库进行搜索由于Cas1 序列在CRISPR-Cas 系统中高度保守[19],且是在CRSIPR-Cas 系统中普遍存在的编码序列,因此根据Cas1 序列进行BLAST 可以有效找出可能含有CRISPR-Cas 系统的基因组。另一种可选的标志序列为CRISPR 序列,CRISPR 序列是CRISPR-Cas 系统中的重要组成部分,因此也可以作为准确识别CRISPR-Cas 系统的序列,为了准确识别CIRSPR 序列,可以选取上述CRISPR 识别软件,通过CRISPR 序列找出的候选序列是通过Cas1 进行序列筛选的2 倍[20],这说明很多CRISPR-Cas 系统是缺乏适应模块的。
1.2.2 筛选对BLAST 识别出的Cas1 序列或CRISPR 识别软件识别出的CRISPR 序列的上下游进行分析,寻找是否有其他编码Cas 蛋白。使用GeneMark 软件中MetaGeneMark_v1.mod 模型[20]对序列进行开放阅读框识别[21]。
对于以Cas1 为标志序列识别出的序列,通过CRISPR-Cas 分类标准来检查其上下游是否存在其他的Cas基因[22]。对于以CRISPR 为标志识别出的序列,在识别出的CRISPR 序列的上下游20 kb[23](有些研究是10 kb[23])以内识别可能编码蛋白的序列。由于Cas9 蛋白和Cpf1 蛋白都由大于1 000 个氨基酸构成[24−26],所以选择氨基酸残基大于500 的编码序列(有些研究是大于700 aa[23]或750 aa[5]作为新Cas 蛋白的候选序列进行下一步分析)。
确定新Cas 蛋白与标志序列和CRISPR 的位置关系,新Cas 蛋白需要在标志序列(Cas1)的4 个基因以内。多数的CRISPR-Cas 系统中Cas 蛋白与CRISPR 序列共同出现的频率很高,限制新的Cas 蛋白至少有50%[23]或70%[5]位于CRISPR 序列上下游20 kb 以内。
2 对识别出的Cas 蛋白序列和CRISPR 序列进行进一步分析
在发掘出新的CRISPR-Cas 系统后,需要对其进行生物信息学分析,以便了解Cas 蛋白的理化性质并进行家族分析。对CRISPR 序列进行分析可以了解该系统对抗的入侵质粒噬菌体等,并有助于研究其切割位点。对识别出的Cas 蛋白序列和CRISPR 序列进行分析流程见图2。
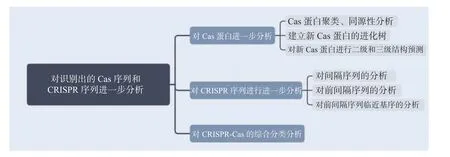
图2 对识别出的Cas 蛋白序列和CRISPR 序列进行分析流程示意图Fig.2 Schematic diagram of the analysis process of the identified Cas protein sequence and CRISPR sequence
2.1 对Cas 蛋白的进一步分析
2.1.1 Cas 蛋白聚类、同源性分析对识别出的蛋白进行聚类分析,聚类分析的目的是根据已有的蛋白序列分析预测新蛋白质序列[27],并对研究蛋白质的起源和家族分析有重要意义[28],将Orthomcl[29]和MCL[30]或作为新Cas 蛋白的聚类分析软件。
为了去除基因组中可能造成偏差的聚类序列,对预测出的Cas 蛋白分析,通过发掘出的Cas 序列和PSI-BLAST 软件[31]对NCBI 的非冗余(nr)蛋白和宏基因组(env_nr)蛋白数据库进行检索,利用HMM 对UniProt 数据库进行检索[32]可得到已知的其他同源蛋白序列[5]。使用HH-suite[33]的HHpred 对发掘出的Cas 蛋白进行远距离的同源蛋白检索,要求新的Cas 蛋白能够检索出10 个同源效应子[23]。
2.1.2 对发掘出的Cas 蛋白进行进化树建立对发掘出的Cas 蛋白和搜索出的同源蛋白建立进化树,比较不同Cas 蛋白之间亲缘关系,是分析新发掘出的Cas 蛋白的常用分析方法之一。通常进化树建立使用软件有RaxmL[34]和PhyML[35]等,上述建立进化树软件输入文件为PHYLIP(.phy)格式。再使用FigTree和iTOL[36]软件实现进化树的可视化。
2.1.3 对预测出的蛋白进行结构域和三级结构预测为了进一步发掘出Cas 蛋白序列特点,进行结构和结构域的预测分析。由于Cas 蛋白进化速度很快,要求识别Cas 蛋白结构域的软件必须能进行精确识别[19,37]。对发掘出的Cas 蛋白进行二级结构预测可以使用JPred4[38]、CD-Search[39]或HH pred[40]。蛋白质的三级结构预测软件分为同源建模法与穿线法,同源建模法预测的原理为相似的氨基酸序列对应着相似的蛋白质结构,如软件Phyre2[41]。穿线法预测通过已知蛋白的结构拓扑进行预测,不相似的蛋白也能有相似的结构,如软件I-TASSER[42]。
2.2 对CRISPR 序列进行分析
2.2.1 间隔序列(spacers)的识别识别CRISPR 序列中的间隔序列(spacers)有助于寻找对抗入侵的质粒和噬菌体。识别CRISPR 序列的CRISPRFinder 等软件识别出的间隔序列根据组装基因组数据确定。相关样品的短DNA 或宏基因组识别间隔序列可使用CRASS 软件[43]。
2.2.2 前间隔序列(protospacer)分析前间隔序列作为CRISPR-Cas 系统进行序列切割在噬菌体或质粒上与间隔序列对应的靶序列,对前间隔序列的识别要求高相似度。查找噬菌体或质粒中的前间隔序列多使用BLAST 软件中的blastn 程序。针对宏基因组数据使用task blastn-short 程序[5]对宏基因组组装数据库进行前间隔序列识别,要求与间隔序列(spacer)的错配碱基小于等于1,对于搜索中可能出现的CRISPR 序列中的间隔序列干扰,通过其重复性去除。除此之外,还可使用megablastn[44]程序,对病毒的非冗余数据库和原核生物基因组数据库进行搜索。此方法要求前间隔序列与间隔序列长度L 最大错配数限制在区间(0,√l−22)[20]。
2.2.3 前间隔序列临近基序(PAMs)分析前间隔序列临近基序(PAMs),是一些Class2 CRISPRCas 系统,如Cas9 蛋白识别靶序列的识别位点,通常在靶DNA 的3′末端作用,有研究猜测PAMs 与DNA解旋作用有关[45]。PAMs 的识别通过前间隔序列侧翼序列的对齐区域进行查找,PAMs 的可视化和DNA图形展示通过WebLogo[46]软件进行。在前间隔序列和间隔序列的对齐过程中,如果出现一个间隔序列与多个不同位置的具有不同侧翼序列前间隔序列匹配,则前间隔序列和下游核苷酸的每一种不同组合都应考虑进PAMs 的计算中[5]。
3 对CRISPR-Cas 系统的分类分析
为了准确分析发掘出的CRISPR-Cas 系统和新的Cas 蛋白,在对其进行进一步分析前,应根据新的CRISPR-Cas 系统进行分类,CRISPR-Cas 系统分类可根据近期发表的分类方法进行[22],根据不同type 和subtype 的标志基因,如Cas3、Cas9和Cas12等对识别出的>500 aa 的CRISPR-Cas 系统进行分类。
CRISPR-Cas 系统分类方法有根据获得模块(Cas1-Cas2)进行分类、根据CRISPR 的序列相似性或结构相似性进行分类、根据Cas1 发生进行分类、根据CRISPR-Cas 基因座结构分类、根据效应模块进行分类、根据亚型分类、根据物种分类。MAKAROVA 等2015 年的研究对比了不同CRISPRCas 系统分类方法的不同(图3),结果表明,通过效应模块进行CRISPR-Cas 系统分类通过蛋白质的相似性能在聚类处理后的库中搜寻到更多结果,通常能够直接对应已经发现的各种亚型[22]。因此,MAKAROVA 等人基于效应模块建立了一种CIRSPR-Cas 系统的自动注释的方法。Cas1-Cas2组成的获得模块作为最普遍的序列未被选择的原因是其虽与Cas1 系统发育树密切相关,但与CRISPR-Cas 基因座结构相关性弱。他们建立的这种分类方法的精确度能达到0.998。
CRISPR-Cas 系统分为两大类(图4):一类(Class1)是多个Cas 蛋白与crRNA 共同作用切割把链的CRISPR-Cas 系统,另一类(Class2)是以Cas9 为代表的单亚基与crRNA 共同作用切割靶链的作用系统。目前的分类方法根据不同的特征基因将Cas 蛋白分为6 种类型,其中Class1 分为3 种类型,Type Ⅰ:以Cas3 或Cas3 基因的变异体为标志基因,在细菌和古细菌中都有广泛分布;Type Ⅲ:以Cas10基因为标志基因,编码多亚基蛋白并包含一个RNA 识别区域,Type Ⅲ在细菌和古菌中也都有分布;Type Ⅳ,缺少编码Cas1-Cas2基因,且部分编码蛋白远离CRISPR 序列,此种蛋白多分布于细菌中。Class2 分为3 种类型:Type Ⅱ:以Cas9基因为标志基因,在细菌和古菌中都有分布;Type Ⅴ:以Cas12和Cas14基因为标志基因,临近Cas1-Cas2 和CRISPR 序列,并与TnpB 蛋白有高度相似性,此种类型大多数分布于细菌中;Type Ⅵ:以Cas13位标志基因[22,47]。

图3 CRISPR-Cas 系统不同分类方法的比较[22]Fig.3 Comparison of different classification methods of CRISPR-Cassystem[22]
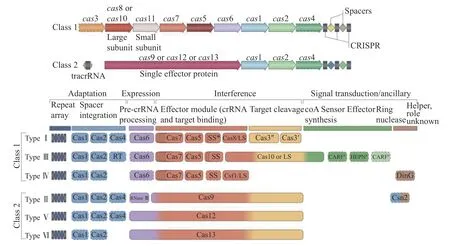
图4 CRISPR-Cas 系统分类图[47]Fig.4 Diagram of classification of CRISPR-Cas systems[47]
新的分类和命名方法根据序列相似性、同源序列分析和上下游序列比较进行CRISPR-Cas 系统的分类。Class2 中,包含了种类Ⅱ、种类Ⅴ和种类Ⅵ及他们的变异体(最新分类),其中type Ⅱ的Cas9 蛋白包含HNH 和RuvC-like 两种结构域,分别切割靶DNA 的两条链。typeV 的Cas12 蛋白只包含RuvClike 结构域负责切割DNA 的两条链。TypeⅥ的Cas13 蛋白包含2 个HEPN 结构域,除此之外,还有非特异性的核糖核酸酶活性。
4 总结与展望
笔者以生物信息学手段为重点,将基于微生物基因组CRISPR-Cas 系统发掘细分为:1)基于隐马尔科夫模型的发掘方法:i)开放阅读框预测,ii)收集已知的Cas 蛋白建立隐马尔科夫模型,iii)CRISPR 序列识别;2)以Cas1 和CRISPR 为标志序列进行CRRISPR-Cas 发掘:i)通过标志序列Cas1 或CRISPR 序列对基因组进行检索,ii)对标志序列的上下游蛋白进行分析寻找可能存在的Cas 酶。提供了在识别出新CRISPR-Cas 系统后,对新CRISPR-Cas 系统的Cas 酶进行的聚类分析(BLAST、HHpred 等软件)、进化树建立(RAxml 等软件)、结构域和三级结构预测(JPred4 等软件)分析方法;3)对新CRISPR-Cas 系统中,CRISPR 序列的间隔序列(CRASS 等软件)、前间隔序列(blastn 等)前间隔序列临近基序分析。
然而,不同的分析方法在实践应用中会有相应的限制。Cas 酶发掘方面,通过隐马尔科夫建立模型的手段只能根据已知的Cas 酶预测出与已知相似的类型,不能预测出序列差别大的两种不同类型Cas 蛋白。通过标志序列Cas1 和CRISPR 序列进行的新Cas 酶发掘对CRISPR-Cas 系统的结构有严格要求,发掘出的CRISPR-Cas 系统必须在上下游20 kb 以内含有标志序列。随着新发现的Class2 CRISPR-Cas14中Cas 蛋白只有400~700 个氨基酸[6],传统认为,单个蛋白可以产生靶向切割作用的Cas 蛋白需要大于950 个氨基酸残基的观点被颠覆,因此,对于标志基因上下游>700 氨基酸残基的蛋白筛选限制条件应当更新。此外,Cas 蛋白进化分类方面随着Cas12 发现可能与TnpB 蛋白转座有关,提供了不同Cas 蛋白起源不同的新观点。CRISPR 序列识别方面,有些软件并不能展示出DR 序列或是序列方向,因此,可能会造成PAM 分析和结构分析的误差。
CRISPR 系统分类上看,随着近年来CRISPR-Cas 系统研究的不断发展,分类方法应不断更新。主要原因如下:1)随着CRISPR-Cas 生物信息学发掘工具的不断发展,已经发现靶RNA 切割的Ⅵ型和Ⅴ型CRISPR-Cas 系统,并有个Ⅴ型的亚型被发现。有研究表明,Ⅴ型CRISPR-Cas 系统是从转座子TnpB 核酸酶通过基因座转移和重复进化产生,因此Ⅴ型CRISPR-Cas 系统出现了大量的突变体,并且有相当一部分进化成了独立的亚型[48]。2)近年来发现的CRISPR-Cas 系统中,被认为在细菌或古菌中执行不同于获得性免疫的功能[49],不含有靶链切割的能力,这些被认为功能不同的CRISPR-Cas 序列通常编码于转座子等可以动的编码区中[48,50]。3)多种涉及到CRISPR-Cas 系统的标志基因与信号传递和调控作用有关[51−52]。
CRISPR-Cas 系统作为定向基因编辑的革命性技术,拥有巨大的潜力和广阔的研究前景。已经发现的Class2 CRISPR-Cas 系统可以定向切割靶单链DNA/RNA 和靶双链DNA,然而,至今为止尚未有科学家发现可切割双链RNA 的CRISPR-Cas 系统。随着越来越多的微生物和宏基因组数据被提供、越来越精进的基因组测序以及不断完善的生物信息学方分析法,会有更多的CRISPR-Cas 系统被发现并应用于基因组的定向编辑,帮助人们了解分析动植物基因功能。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
体育教学(2022年4期)2022-05-05
有色金属(矿山部分)(2021年4期)2021-08-30
数学小灵通(1-2年级)(2020年11期)2020-12-28
资源导刊(信息化测绘)(2020年5期)2020-06-22
启蒙(3-7岁)(2019年5期)2019-06-27
小学生学习指导(低年级)(2019年3期)2019-04-22
漫画月刊·哈版(2019年4期)2019-04-18
中国卫生(2015年10期)2015-11-10
读写算·小学低年级(2014年4期)2014-07-24
