
河南省部分区域肾综合征出血热患者汉坦病毒的分子特征
2021-04-14 09:20:48郑礼钧李三景霍玉奇
中国人兽共患病学报 2021年3期
马 杰,郑礼钧,唐 静,李三景,霍玉奇
汉坦病毒(Hantavirus, HV)属于布尼亚病毒目(Bunyavirales)、汉坦病毒科(Hantaviridae)、正汉坦病毒属(Orthohantavirus),是分布最广泛的人兽共患病病毒之一[1-2]。汉坦病毒是一种分节段的单股负链RNA病毒,其基因组有大(L)、中(M)、小(S)3个片段组成,依次编码病毒RNA依赖的RNA聚合酶、包膜糖蛋白(Gc和Gn)以及核衣壳蛋白(NP)[2-4]。HV主要通过吸入被感染啮齿动物尿液和粪便产生的气溶胶,进食病毒污染的食物,接触被感染啮齿动物唾液传播给人类,鲜有因被啮齿动物咬伤而被传染的报道[5-6]。尽管HV在其天然宿主啮齿动物中不会引起明显的疾病,但HV可能在其机会和终末宿主人类中导致两种严重的综合症,一种是由旧世界HV引起的肾综合征出血热(hemorrhagic fever with renal syndrome,HFRS),例如亚洲的汉滩病毒(hantaan virus, HTNV)和汉城病毒(seoul virus, SEOV),欧洲的普马拉病毒(puumala virus, PUUV)和多布拉法-贝尔格莱德病毒(dobrava-belgrade virus, DOBV)等,一种是由新世界HV引起的HV心肺综合征(Hantavirus cardiopulmonary syndrome,HCPS),例如美洲的辛诺柏病毒(sin nombre virus, SNV)和安德斯病毒(andes virus, ANDV)等[7-8]。
我国自1955年以来一直将HFRS视为重要的公共卫生事件[9]。我国主要以HTNV和SEOV为主要流行株[10]。自1963年河南省驻马店正阳县首次报告HFRS病例以来,HFRS对河南省的公共卫生构成的威胁持续上升。但目前河南省未见从患者中成功分离到HV的报道,一直缺少HFRS患者HV分离株基因分型或基因亚型的分布和分子生物学特征分析研究。为此本研究通过提取HFRS患者血清中HV RNA并扩增S片段和M部分片段核苷酸序列,与已分离到的国内其他相对应的基因序列进行同源分析和构建系统发生树,明确本研究中HV分离株的基因型别和分子特征。
1 材料与方法
1.1标本来源 2017-2018年就诊于河南省郑州市第六人民医院且临床诊断为HFRS的患者血清标本32份。
1.2病毒RNA提取 采用TaKaRa Mini BEST Viral RNA/DNA Extraction Kit Ver.5.0核酸提取试剂盒(宝生物工程大连有限公司),按步骤提取病毒RNA。
1.3引物的设计及合成 参考文献[11-12],并根据GenBank数据库中HV基因序列,设计5组半巢式引物。3组扩增HTNV S片段的核苷酸序列:HTNV-S1F/HTNV-S740R/HTNV-S1160R、HTNV-S357F/HTNV-S595F/HTNV-S1160R和HTNV-S357F/HTNV-S934F/HTNV-S1702R;1组扩增SEOV 部分S片段核苷酸序列:SEOV-S1F/SEOV-S207F/SEOV-S963R;1组扩增HTNV M部分片段核苷酸序列:HTNV-M55F/HTNV-M107F/HTNV-M939R(表1)。

表1 S和M部分片段序列半巢式PCR扩增引物引物详细信息Tab.1 S and partial M segment sequences of primers used for hemi-nested PCR
1.4半巢式PCR和测序 以上述血清标本中提取的RNA为模板,采用RevertAid first strand cDNA synthesis kit(赛默飞世尔科技有限公司)按说明书合成cDNA。先分别用上述引物HTNV-S357F/HTNV-S595F/HTNV-S1160R和SEOV-S1F/SEOV-S207F/SEOV-S963R扩增S部分片段序列,确定阳性样本。然后再用其他引物扩增相应片段序列。PCR分两轮进行,反应条件相同,具体为94 ℃预变性4 min,94 ℃ 30 s,55 ℃ 30 s,72 ℃ 60 s,共35个循环,72 ℃延伸10 min;扩增产物用1%琼脂糖凝胶电泳,若扩增条带大小与预期片段大小相同,则表明为特异性扩增产物。扩增产物送生工生物工程(上海)股份有限公司进行测序。
1.5核苷酸序列分析 采用Contig Express软件对S片段序列进行校对和拼接,应用DNAStar软件内置的Editseq 程序对S片段和M部分片段核苷酸序列进行编辑,使用该软件中Megalign程序对核苷酸序列进行同源性分析。采用MEGA X软件用邻位相连法(neighbor-joining, NJ)分别对S片段和M部分片段核苷酸序列构建进化树。它们对应的最优模型分别是GTR+G和T92+G,bootstrap值统一设置为1 000次重复。用于构建进化树的序列均来自GenBank。
2 结 果
2.1阳性标本筛检及分型 从收集到的32份血清标本提取RNA,反转录成cDNA后,分别用HTNV-S357F/HTNV-S595F/HTNV-S1160R和SEOV-S1F/SEOV-S207F/SEOV-S963R引物,经半巢式PCR扩增,其中16份标本扩增出特异性条带。对扩增条带进行回收和测序,结果表明16位患者均是HTNV引起的HFRS。16位HTNV患者主要分布在许昌5例,平顶山2例,漯河2例,周口3例,驻马店2例,商丘1例,濮阳1例。
2.2S片段和M部分片段的核苷酸序列编辑 用HTNV特异性引物扩增S片段和M部分片段核苷酸序列。经测序,序列拼接和校正后,分别得到16株S片段序列和16株M部分片段序列。结果表明16位患者均为HTNV阳性患者。S片段的长度为1 701 bp或1 702 bp;M部分片段的长度为732 bp。整理后的序列递交NCBI GenBank数据库,S片段和M部分片段的登录号分别为MN478382-MN478397 和MN478398-MN478413。
2.3同源性分析 分别用S片段和M部分片段序列对16株HTNV进行同源性分析。16株HTNV中的HN2018/138株与其余15株相比核苷酸序列差异较大,同源性分别为91.4%~92.1%(S片段)和83.2%~84.4%(M部分片段);其余15株HTNV同源性高,分别为97.1%~100%(S片段)和96.9%~99.9%(M部分片段)。HN2018/138株的核苷酸序列经基于局部比对搜索工具(Basic Local Alignment Search Tool, BLAST)检索后,发现其S片段序列与陕西的SXRn20120013株和XAAa10091712株最相近(>98.1%),其M部分片段序列与陕西的XAAa10091712株和贵州的Q32株最相近(97.8%)。
2.4系统发生分析 利用S片段核苷酸序列构建系统进化树,16个分离株聚集成簇,从系统发生树上可以看出其中15株亲缘关系较近,分布较集中,而HN2018/138株单独形成一个分支,它与陕西和贵州的病毒株亲缘关系较近。HN2018/138株为S2亚型,其他15株病毒分离株为S5亚型。各分离株成簇情况在基于M部分片段构建的系统发生树与基于S片段构建的系统发生树上基本保持一致(图1、2)。
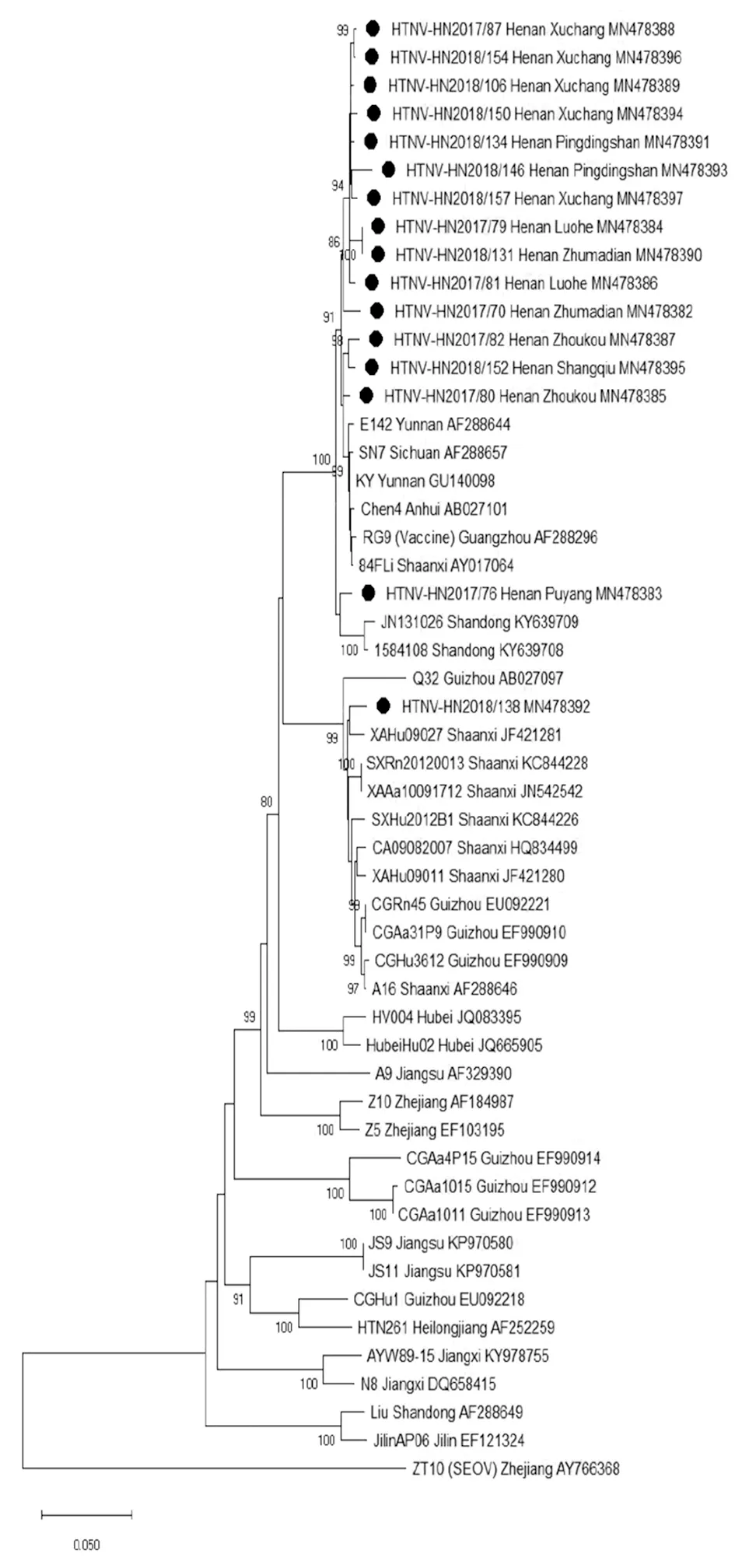
图1 S片段核苷酸系统发生分析Fig.1 Phylogenetic tree of HV based on complete S segments
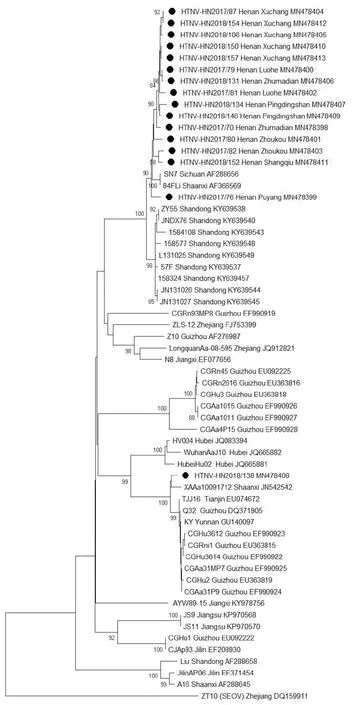
图2 部分M片段核苷酸系统发生分析Fig.2 Phylogenetic tree of HV based on partial M segments
3 讨 论
流行性出血热是我国法定乙类传染病,又称肾综合征出血热(HFRS)。自1981年实施综合预防措施以来,包括河南省在内的一些省份的HFRS发病率已大大降低[13]。然而,近些年河南省HFRS患者人数激增,从2014年的138例增加到2018的416例。我国通过法定传染病上报系统上报的HFRS病例未对HTNV或SEOV感染进行区分[5]。近些年研究人员从流行特征方面对河南省许昌、驻马店、周口等地区的HFRS患者进行了分析。本研究从病毒基因入手,利用半巢式PCR技术,分别用HTNV和SEOV特异性引物,从16份血清标本中成功扩增出病毒基因组片段,经序列分析均为HTNV,无SEOV。系统发生树分析也证实16株病毒株为HTNV。
河南省是我国HFRS的主要疫区之一,也是我国最早发现SEOV的地区。根据流行性出血热监测及流行病学研究结果表明,河南省存在着HTNV和SEOV两种病毒株。20世纪90年代河南省主要的HFRS疫区已被证实存在单纯的HTNV型、SEOV型和混合型疫区3种。孙黎等2005年对河南省安阳、开封、驻马店、南阳等7个HFRS高发地区捕获的啮齿类动物检测后发现褐家鼠、黄胸鼠和小家鼠携带的病毒均为SEOV[14]。杜燕华等对驻马店确山县捕获的啮齿类动物检测后发现其携带的病毒均为SEOV[15]。有研究证实在法国捕获的褐家鼠中持续检测到SEOV,但在欧洲没有人感染SEOV的报道[16]。本研究中分离到的病毒株型别与前人不一致,可能原因有:1)前人研究的是HV宿主动物;2)本研究的样本量太小,并不能反映河南全境的真实情况;3)因为由SEOV引发HFRS疾病的严重程度比由HTNV引发HFRS疾病的严重程度要轻[17-18],造成SEOV引发的HFRS患者未入院治疗或就近入院治愈。这些原因可能导致本研究从HFRS患者血清中只检测到HTNV。
16位HTNV阳性HFRS患者主要集中在河南省中部地区。16株HTNV中15株病毒株在S片段和M部分片段的核苷酸序列同源性都大于96%, 而HN2018/138株与其余15株同源性为91.4%~92.1%(S片段)和83.2%~84.4%(M部分片段)。根据前人的分子流行病学研究结果,我国流行的HTNV至少可分为9个亚型[13]。HN2018/138株属于S2亚型,与其他15株不同(S5)。而同地区HV分离株倾向于聚集在同一分支,有明显的地域聚集性[19-20]。本研究中样本编号为HN2018/146的患者入院20 d前从平顶山叶县到山东务工,15 d前开始发热。根据HTNV感染在突发高热等临床症状之前,通常有大约 2~4周的潜伏期[21]。该患者可能在平顶山叶县感染了HTNV。经BLAST分析发现HN2018/146株与来自山东省的分离株之间的同源性低于96%,而与来自平顶山鲁山HN2018/134分离株之间的同源性高于98%。系统发生树显示HN2018/146株与HN2018/138株外的其他分离株在同一分支。因此样本编号为HN2018/146的患者在平顶山感染HTNV的可能性较大。样本编号为HN2018/138的患者 1个月前从河南省周口市到江苏省南京市务工,5 d前开始发热。经序列比对发现HN2018/138株与来自江苏省的分离株之间的同源性为86.8%~89.5%(S片段)和82.8%(M部分片段),差异较大。进一步与周口市分离株HN2017/80株和HN2017/82株进行序列比对发现,它们的同源性为92.0%~92.2%(S片段)和83.3%~84.4%(M部分片段),差异也较大,但S片段的同源性相对较高。同样在驻马店分离的两株序列的同源性为高于98%(S片段和M部分片段);在许昌分离的5株序列的同源性高于99%(S片段和M部分片段)。通过GenBank数据库经Blast后,发现HN2018/138株S片段序列同源性与陕西的SXRn20120013株和XAAa10091712株最相近(>98.1%),其M部分片段同源性与陕西的XAAa10091712株和贵州的Q32株最相近(97.8%)。系统发生树分析同样表明HN2018/138株与自陕西和贵州的病毒株在同一分支,但样本编号为HN2018/138的患者未曾去过陕西和贵州。因为GenBank数据库中河南和江苏可用的HTNV序列较少,推测HN2018/138株可能为本地变异株,或省外输入病毒株。
利益冲突:无
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04 11:40:51
生物学通报(2022年1期)2022-11-22 08:12:18
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
肝博士(2022年3期)2022-06-30 02:48:28
中学生数理化·中考版(2022年5期)2022-06-05 07:52:38
中学生数理化·七年级数学人教版(2022年5期)2022-06-05 07:51:56
中学生数理化·七年级数学人教版(2022年6期)2022-06-05 06:51:00
中学生数理化·八年级物理人教版(2022年4期)2022-04-26 14:11:18
南京林业大学学报(自然科学版)(2021年5期)2021-10-13 02:06:16
Journal of Sport and Health Science(2019年6期)2019-11-26 07:30:53
