
2006-2016年贵州省炭疽芽胞杆菌SNR分型分析
2021-04-14 09:20:50姚光海李世军
中国人兽共患病学报 2021年3期
马 青,杨 平,刘 英,王 月,聂 炜,姚光海,李世军
炭疽是一种自然疫源性疾病,具有全球流行性。炭疽芽胞杆菌是炭疽的病原体,属于需氧芽胞杆菌属细菌,可感染各种家畜、野生动物及人类。由于其病原体的环境耐受性和感染破坏性等特点,炭疽无法根除。人类主要通过接触炭疽病畜毛皮和食肉而感染,也可以通过吸入含有炭疽芽胞的粉尘或气溶胶而感染,主要表现为皮肤炭疽,其次为肺炭疽、肠炭疽和脑膜炎炭疽[1]。
炭疽芽胞杆菌在遗传过程中,基因组上存在能稳定遗传的单核苷酸突变位点,单核苷酸重复序列分析(Single-Nucleotide Repeat Analysis, SNR)通过分析不同分离株之间等位基因上单碱基的重复数,可区分同一MLVA型的近缘分离株,从而更细致的反映细菌的遗传进化差异,SNR因此成为分子流行病学调查的有效技术手段,近年来被广泛用于炭疽杆菌分型研究[2]。本研究采用SNR方法对来自贵州省2006-2016年间不同地区的炭疽芽胞杆菌进行SNR分析,探讨其SNR型别分布特征,为炭疽疫情监测、调查与溯源提供技术手段和科学依据。
1 材料与方法
1.1菌株来源 本研究所用菌株为本实验室2006-2016年间收集保存的贵州省各地区炭疽芽胞杆菌,共71株。其中5株来自患者标本(4株分离自皮肤渗出物,1株分离自全血),3株来自动物标本(狗肝、狗脾、病死牛肺各分离1株),63株来自外环境标本(2株分离自牲畜饮用水, 61株分离自土壤)。
1.2主要试剂 血琼脂平板购自郑州博赛公司,Taqmix聚合酶、灭菌去离子水、DNAMarker、琼脂糖均购自大连宝生物工程有限公司。
1.3 方 法
1.3.1炭疽杆菌基因组DNA的提取 将炭疽菌株接种于血平板培养18~24 h,挑取单菌落于0.2 mL无菌纯水中制成菌悬液,100 ℃加热 10 min,10 000 r/min离心 10 min,取上清液经0.22 μm滤器过滤后备用。
1.3.2SNR位点引物设计 采用文献[3]报道的4个SNR多态性位点(CL10、CL12、CL33、CL35)及其引物序列进行SNR分析,引物序列委托北京天一辉远生物科技公司进行引物合成。详细引物序列信息见表1。

表1 引物序列信息Tab.1 Information on primer sequences
1.3.3PCR扩增 采用25 μL反应体系进行扩增,体系包含Taqmix12.5 μL,上下游引物各1 μL,DNA 模板1 μL,去离子水9.5 μL。扩增参数为:94 ℃变性2 min;94℃ 30 s、45℃ (CL12)30 s/ 50℃ (CL35)30 s/55℃ (CL10和CL33)30 s(各位点引物退火温度不同)、72℃ 30 s,40个循环;72℃延伸10 min。
1.3.4毛细管电泳 将扩增产物经琼脂糖凝胶电泳确认为清晰单一条带后,委托北京天一辉远生物公司进行毛细管电泳分析。本研究用3730xl测序仪进行毛细管电泳,得到原始图谱(fsa文件),使用GeneMapper v4.0软件分析后生成PDF图谱和Excel文档(包括size、基因分型等信息)。为了保证结果的准确,实验过程中要作以下质控:①对相同样本进行3次实验(2次重复)并获得一致的结果。②STR检测结果峰值需达到400RFU,否则该标本必须重新进行检测;③分析前检查所有标本内标是否正确;④每次实验必须同时做阴性对照和质控阳性对照。
1.3.5SNR数据聚类分析 根据毛细管电泳所得本次所测71株菌株各SNR位点的序列长度,运用Bionumerics7.6软件进行聚类分析,获得SNR型别及构建最小间距图。
2 结 果
2.1炭疽芽胞杆菌SNR位点序列长度 71株贵州省炭疽芽胞杆菌菌株基因组DNA经各SNR位点(CL10、CL12、CL33、CL35)引物扩增和毛细管电泳,获得了菌株各SNR 位点的序列长度(表 2)。
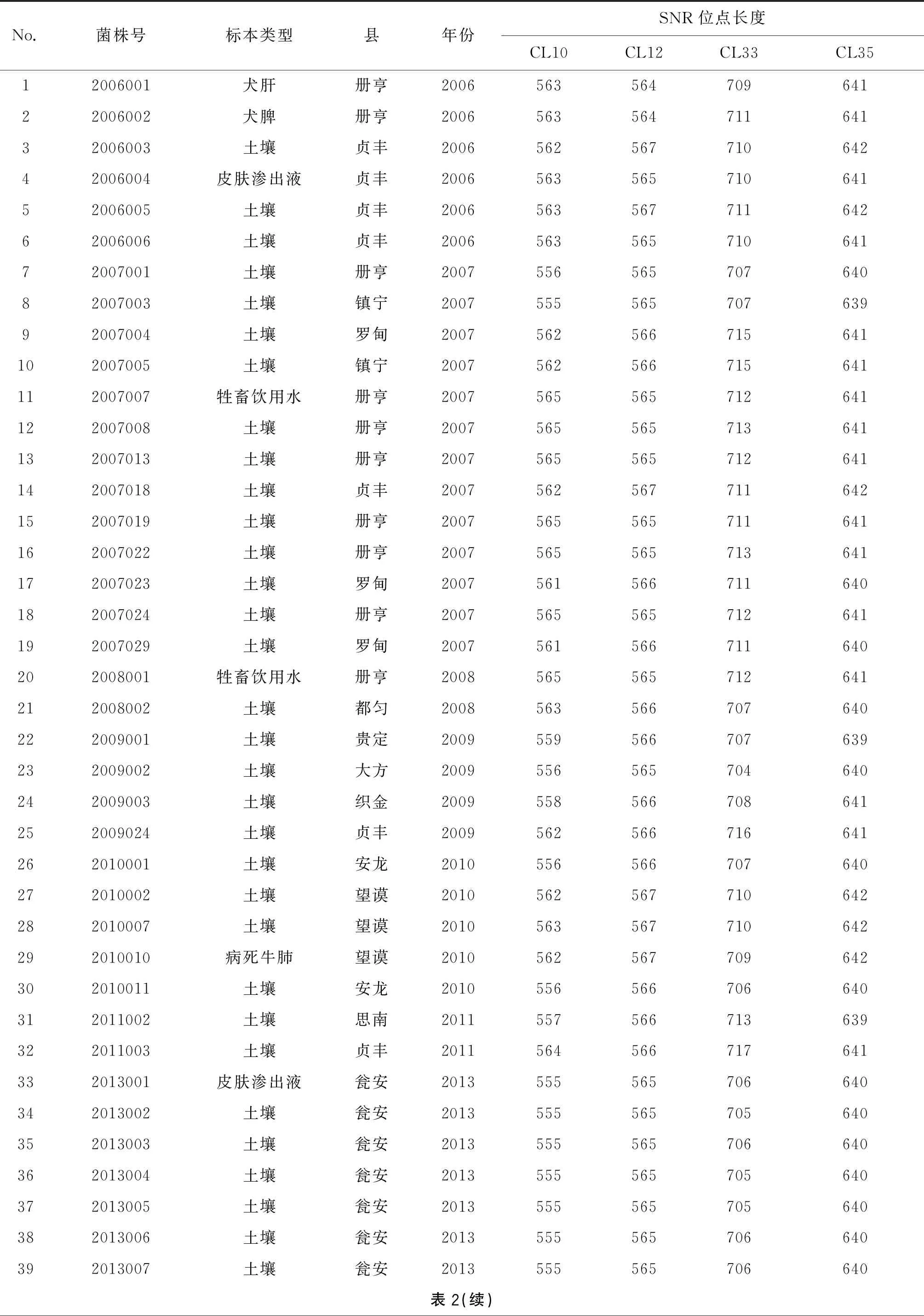
表2 71株炭疽杆菌SNR位点序列长度Tab.2 Sequence lengths for SNRs of 71 Bacillus anthracis strains
2.2炭疽芽胞杆菌SNR数据聚类分析结果 基于SNR各位点序列长度的聚类分析显示(图1),按照4个SNR位点基因完全相同定义为一个基因型,可以将71株菌分为37个SNR型,聚类图中71株菌可分为A和B两群,A群可进一步分为A1和A2亚群,B群可进一步分为B1和B2亚群,各亚群又可分为若干分支,其中A群有27个分支,B群有10个分支。71株菌中有26株分型为A2a,占全部菌株的36.62%,其次为A1a型,共有16株,占全部菌株的22.54%。菌株的聚类关系具有一定的地域性和时间聚集性。
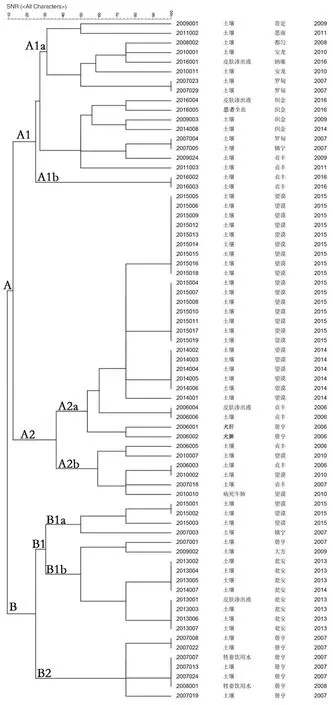
图1 71株炭疽杆菌SNR聚类分析Fig.1 Cluster analysis based on SNR data for 71 Bacillus anthracis strains
2.3炭疽芽胞杆菌SNR最小间距图分析结果 基于炭疽芽胞杆菌SNR各位点序列长度的最小间距图显示,71株菌株被分别为7个SNR克隆群和6个单一的SNR型。大部分相同或相邻区(县)遗传距离相对较近,但同时也存在不同区(县)或年份来源菌株距离较近而相同县来源菌株距离相隔较远的现象(图2)。
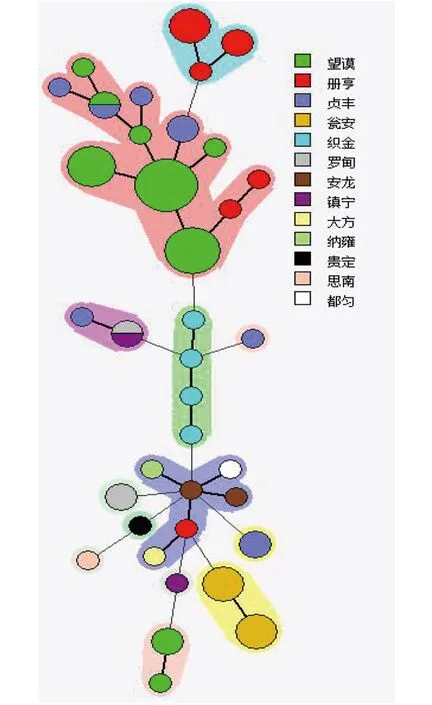
图2 71株炭疽杆菌SNR分型最小间距图Fig.2 Minimum spanning tree based on SNR data for 71 Bacillus anthracis strains
3 讨 论
贵州省炭疽疫源地分布广泛,随着居民生活水平的提高,近年来病例虽有所减少,但人间炭疽疫情时有发生,疫情高于全国平均水平[4-6]。由于炭疽杆菌可形成芽胞在疫源地土壤环境中可存活长达数十年,疫源地存在炭疽流行的风险,因此了解当地炭疽芽胞杆菌的分子型别特征对预防和控制当地炭疽疫情具有重要意义。
炭疽菌株遗传信息具有极端的同源性,现有研究证明炭疽杆菌缺少分子的多样性,质粒电泳没有差异,基因组AFLP、rRNA基因RFLP和保护性抗原的DNA序列差异非常小[7-8]。常用的脉冲场凝胶电泳(PFGE)分子分型技术,随机扩增多态性DNA标记(RAPD)技术仅能将炭疽芽胞杆菌同蜡状芽胞杆菌和枯草芽胞杆菌区分开来,即仅能鉴定到种属间的差异[9-10]。多位点可变数量串联重复序列分析(MLVA)方法被证明可用于鉴别炭疽菌株之间的差异及多样性,是较好的分子分型方法之一,但仅能将不同的炭疽芽胞杆菌分离成不同的大基因组。单核苷酸多态性(SNP)分析通过直接比较不同分离株基因组序列,找出单个碱基的差异,但也仅能分辨相似分离株,且操作繁琐[2]。从自然暴发炭疽疫情和生物恐怖袭击事件中获得的炭疽分离株往往具有极低的基因多样性,在这种需要区分遗传进化关系较近的分离株的情况下,MLVA 和 SNP 就不能满足要求[2]。单核苷酸重复序列分析(SNR)是近年建立的分辨率更高的基因分型方法,Beyer等[3]选取了CL10、CL12、CL33和CL35共4个位点对384株分离菌进行了研究,有效区分了同一MLVA基因型的不同菌株。
本研究利用4个SNR位点的单核苷酸重复序列分型方法将2006-2016年贵州省分离获得的71株炭疽芽胞杆菌进行SNR分型分析,了解贵州省近年来炭疽芽胞杆菌的SNR型别特征。SNR分析结果显示,71株炭疽芽胞杆菌被分为37个SNR基因型。本文的聚类分析显示,71株菌株可分为A和B两群,A群可进一步分为A1和A2亚群,B群可进一步分为B1和B2亚群,各亚群又可分为若干分支,提示贵州省炭疽芽胞杆菌具有SNR型别多样性。2006001和2006002菌株来源于同一只犬,被分为A2a亚群的同一基因型。71株菌株的聚类图和最小间距图均显示菌株基因型分布具有一定的地域性和时间聚集性,而与菌株分离来源无明显相关性,提示贵州省不同地区和不同年份间的流行菌株并非来源于同一克隆株。鉴于SNR分型技术具有更高分辨率的特点,SNR还能区分同一地区同一时间(同一疫情)的菌株。聚类分析显示,分离自相同年份相同县份同一疫情(2013年瓮安县)的7株菌株虽聚类较近,仍被分为2个不同的SNR基因型,而我们以往采用MLVA技术分型结果显示这7株菌为同一MLVA基因型[11]。此外,2013年不同来源的2013001、2013003、2013006和2013007菌株聚类较近,2006年来自患者的200604菌株与来自土壤的200606菌株聚类较近,提示来自相同年份和地区(县)的环境(土壤)或动物来源菌株与患者来源菌株聚类较近,反应了当时引起人间炭疽疫情的菌株的可能来源。
综上所述,本研究应用基于4个位点的SNR技术对贵州省2006-2016年间分离的71株炭疽芽胞杆菌进行SNR分型分析,结果提示贵州省近年来的炭疽芽胞杆菌具有SNR型别多样性,且SNR型别分布具有一定的地域和时间聚集性的分布特点,结果反映了不同地区来源菌株的复杂性。通过SNR技术可进一步了解贵州省菌株的遗传与进化关系,揭示贵州省炭疽芽胞杆菌的基因型别和分子流行病学特征,为当地炭疽疫情的预警和防控提供病原学依据。
利益冲突:无
猜你喜欢
现代临床医学(2023年1期)2023-03-24 08:30:06
中国生物防治学报(2022年3期)2022-07-09 10:00:22
中国医药科学(2022年5期)2022-05-05 23:58:07
微生物学杂志(2021年2期)2021-07-01 11:01:06
小学生必读(中年级版)(2021年12期)2021-03-03 07:43:44
小学生必读(中年级版)(2021年11期)2021-02-22 05:19:06
宁夏医学杂志(2020年4期)2021-01-21 08:25:16
微生物学杂志(2020年2期)2020-12-31 07:17:13
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
中国卫生标准管理(2015年3期)2016-01-14 03:41:45
