
着色性干皮病患者的临床及基因突变分析
2021-04-14 05:44韦琴丁艳
世界最新医学信息文摘 2021年18期
韦琴,丁艳
(1.江汉大学医学院,湖北 武汉 430000;2.武汉儿童医院,湖北 武汉 430000)
0 引言
着色性干皮病(Xeroderma Pigmentosum,XP)是一种罕见的常染色体隐性遗传皮肤病,发病率约为1-1.65/10 万,1874 年由皮肤科医生Moriz Kaposi 首次报道[1]。患儿多在出生后6 个月至3 岁发病,临床表现为皮肤对日光特别是紫外线高度敏感,暴露部位皮肤出现色素沉着、干燥、角化、萎缩及癌变[2];由于皮肤癌变和神经退行性后遗症,XP 患者的寿命往往较短。根据DNA 损伤修复缺陷的基因不同,XP 可分为八个基因型,分别为互补组 XPA、XPB、XPC、XPD、XPE、XPF、XPG,以及一个变异型 XPV[3]。其中,XPD 致病基因为 ERCC2,该类型在我国十分罕见,目前国内仅有3 例报道。本研究结合我院诊断的1 个XP家系并复习相关文献,探讨ERCC2 基因导致着色干皮病(XPD)的临床特征,进一步加深临床医生对该疾病的认识。
1 对象与方法
1.1 研究对象
患者,男,6 岁,因全身皮肤色素异常并渐加重5 年6 个月,乏力半月入院,初步诊断“着色性干皮病”。患者自半岁起,面部皮肤出现粟粒至花生米大小的黑褐色色素沉积,部分融合成片,渐延及躯干、四肢等部位,并逐渐增多。体格检查:神清,皮肤色泽暗黄,散在点状色素沉积,部分成片,双下肢可见散在点状出血点,左侧颈部可触及数枚黄豆大小肿大淋巴结,无压痛;心肺听诊及腹部触诊未见异常;语言、运动、智力发育均落后同龄人;神经病理征阴性。实验室检查:WBC 10.51*109/L,HGB 72g/L,PLT 98*109/L;炎症指标高,肝肾功能、心肌酶谱、大小便常规基本正常;肺部CT 示双肺异常密度影,右侧肺门多处钙化灶,怀疑结核。患儿淋巴结穿刺液涂片示荧光染色抗酸杆菌阳性,肺结核诊断明确,予以抗肺结核药物治疗+颈部淋巴结病灶清除术。患儿为第2 胎、顺产,自幼体弱,易反复呼吸道感染,生长发育及智力均落后于正常同龄儿。父母表型正常,患者2 为患者1 的姐姐,12 岁,出生后2 月出现面部黑褐色色素沉着斑,皮肤色素沉着程度更严重。患者父母非近亲结婚;患者的外祖父、外祖母近亲结婚,表型均正常。
1.2 高通量测序检测致病基因
经家属知情同意后取患者、患者姐姐及其父母外周血各2mL,采用外周血基因组DNA 提取试剂盒(Omega 公司)提取患者DNA,并测定DNA 的浓度。采用Illumina 公司Truseq Exome Library 建库试剂盒以及IDT The xGen Exome Research Panel v1.0 全外显子捕获芯片对患者的DNA 进行文库构建,并利用Illumina 高通量测序仪Hisseq2000 进行全外显子组测序。原始数据去除低质量的读长,比对到hg19 参考基因组的reads 经过组装拼接后,采用GATK 以及VarScan 软件进行突变、SNPIndel 的识别、注释并利用SIFT、Polyphen-2 以及Mutation Taster 等生物信息学软件评定突变位点的生物学影响。结合临床表型利Clinvar、HGMD、pubmed 等数据库对给出的突变数据进行再次分析,确定致病位点。利用NCBI 数据库检索突变位点上下游序列并设计引物,利用PCR 技术扩增含有突变位点的片段序列并进行sanger 测序(3500DX 测序仪)验证患者及家属。
1.3 生物信息学分析
对检测出的ERCC2 基因突变利用Mutation Taster、Polyphen2等在线软件预测变异的致病性,利用生物信息学在线软件进行不同物种间EHMT1 蛋白序列的保守性分析,利用ACMG 指南评估突变的临床意义。
2 结果
经过全外显子测序以及Sanger 一代测序验证后发现,患者1 的ERCC2 基因(NM_000400)存在c.1847 G>A 以及c.1988 G>A 复合杂合突变,采用同样的方法患者2 也检测到上述两种突变。c.1847 G>A 与c.1988 G>A 这两个杂合突变分别来源于患者父亲、母亲。
c.1847 G>A 突变遗传来自患者父亲,该突变导致ERCC2基因编码的蛋白第616 位氨基酸由精氨酸突变为谷氨酰胺(p.R616Q),根据ACMG 指南该位点为致病性改变(PS1 与已知的致病变异相同;PM2 文献数据库记录的低频变异;PP1:支持家系共分离,PP3 多种生信软件预测突变对蛋白功能有影响)。
c.1988 G>A 突变来自患者母亲,该突变导致ERCC2 基因编码的蛋白第663 位氨基酸由半胱氨酸突变为络氨酸(p.C663Y),依据美国医学遗传学与基因组学学会(ACMG)指南[4]为可能致病性改变(PM2 低频变异;PM5:变异位于之前报道过的氨基酸残基;PP1:支持家系共分离,PP3 多种生信软件预测突变对蛋白功能有影响),该位点未见报道,氨基酸保守性分析发现该位点在不同的物种间保守,提示该位点异常可能影响蛋白的功能或稳定。
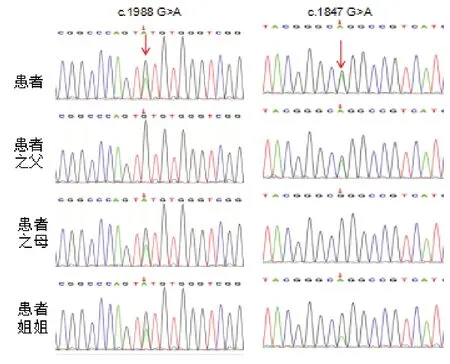
图1 两例患者及其父母ERCC2 测序结果分析
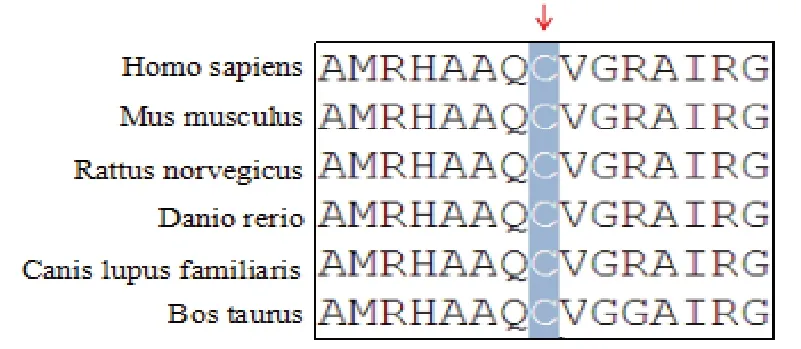
图2 突变位点663 位氨基酸(半胱氨酸)在不同物种间的保守性分析
3 讨论
本研究中的家系患者均在半岁以内发病,患者1 步态不稳,合并肺结核感染可能与母亲曾感染肺结核有关;患者2 皮肤色素沉着更严重,可能与防晒护理较晚有关。经过全外显子测序后发现,该家系成员的ERCC2 基因(NM_000400)存在c.1847 G>A(p.R616Q) 与c.1988 G>A(p.C663Y) 复 合 杂 合 突 变,其 中c.1988 G>A(p.C663Y)为新发突变。
ERCC2 基因位于19 号染色体19q13.32 区域,基因全长21Kb,编码87kD 大小的蛋白,ERCC2 作为DNA 修复基因,是ATP 依赖性的转录因子II(TFIIH)复合物的一部分,可通过5"-3"解旋酶活性分离双螺旋结构[5-7]。研究表明TFIIH 复合物参与核苷酸切除修复途径(NER),该途径可以修复由化学治疗和基础转录引起的DNA 损伤。ERCC2 基因在外源性基因逆转录过程中和内源性基因的逆转录过程中均发挥抑制作用,因此它不仅参与DNA 损伤修复也可能在防御逆转录病毒感染中发挥作用[8]。有实验研究证实,ERCC2 基因缺陷的细胞更容易感染人类免疫缺陷病毒及Moloney 小鼠白血病病毒[9]。虽然ERCC2抑制逆转录的机制尚不清楚,但是ERCC2 保护基因在防止了不良DNA 片段插入方面发挥关键作用,使得基因组保持完整性[10]。
目前文献及数据库中收录了 100 多种与疾病相关的ERCC2 基因突变,其基因型与表型关系复杂;不同类型或者不同位点的突变导致的临床表型及严重程度差异很大,如患者可表现为XPD、XPD 合并 Cockayne 综合征、毛发硫营养障碍以及脑-眼-面-骨骼综合征、膀胱移形细胞肿瘤等[11]。当ERCC2基因突变表现为XPD 时,该基因突变位点通常发在在Rad51 功能域或是RecA-like 功能域,这2 个区域分别又称HD1、HD2,主要负责调节其解旋酶活性[12,13]。本案例中,患者1 和患者2的ERCC2 基因突变位点均在HD2。
XP 亚型在不同的国家和地区具有显著的分布差异,其中我国患者主要是XPA、 XP-C、XP-G、XP-V 这4 种类型,XP-A最为常见,而XP-D 极为罕见[14]。XPD 患者一般起病较早,皮肤色素沉着明显,肿瘤发生率高。部分患者并发神经系统损害,如步态不稳、肌张力低下、肌痉挛等[15]。虽然该家系患者尚未并发皮肤肿瘤和神经系统损害,但应严格避光,定期复查。
综上所述,本研究报道了国内罕见的1 个着色性干皮病D型家系,发现了ERCC2 基因一个未见报道的新突变,丰富了人类遗传性疾病的基因突变数据库。在临床工作中,病史资料结合基因检测,有助于本类疾病的早诊断与精确分型,为患者及家属提供治疗管理和遗传生育方面的建议。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
河北果树(2021年4期)2021-12-02
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
三农资讯半月报(2020年2期)2020-03-09
中南林业科技大学学报(2019年4期)2019-04-08
小学生导刊(2018年13期)2018-06-29
森林工程(2018年1期)2018-05-14
中学生理科应试(2017年6期)2017-09-27
