
莪术醇原药高效液相色谱分析方法研究
2021-04-12 10:59郑杨,李岩,刘博,周芹
农药科学与管理 2021年3期
郑 杨,李 岩,刘 博,周 芹
(1.黑龙江省植检植保站,黑龙江 哈尔滨 150090;2.中国农业大学植物保护学院,北京 100083;3.黑龙江大学农学院,黑龙江 哈尔滨 150080)
1 前言
目前国内对莪术醇原药分析方法尚未见报道。本文采用高效液相色谱法对莪术醇进行定性和定量分析。该方法操作经济、快速、简便、准确,分离效果好,准确度和精密度均能达到定量分析的要求,适用于农药产品质量的检测分析。
2 试验部分
2.1 试剂和溶液 乙腈:色谱纯;甲酸:分析纯;超纯水(电阻率18.2MΩ·cm,25℃);莪术醇标样:99.1%;试样:莪术醇原药。
2.2 仪器 高效液相色谱仪:Agilent1260,具有二级管阵列检测器和自动进样器,定量环20μL;色谱工作站;Millipore超纯水制备系统;色谱柱:250mm×4.6mm(i.d)不锈钢柱,内装Zorbax SB-C18、5μm填充物;过滤器:滤膜孔径约0.45um。
2.3 液相色谱操作条件 流动相:乙腈:0.1%甲酸水溶液=85:15;流速:1.0mL/min;柱温:30℃;检测波长:210nm;进样体积:5.0uL;保留时间:莪术醇5.3min。
2.4 测定步骤
2.4.1 标样溶液的配制 准确称取莪术醇标样0.031g(精确至0.2mg),置于100mL容量瓶中,用乙腈超声溶解并稀释至刻度,静置备用。
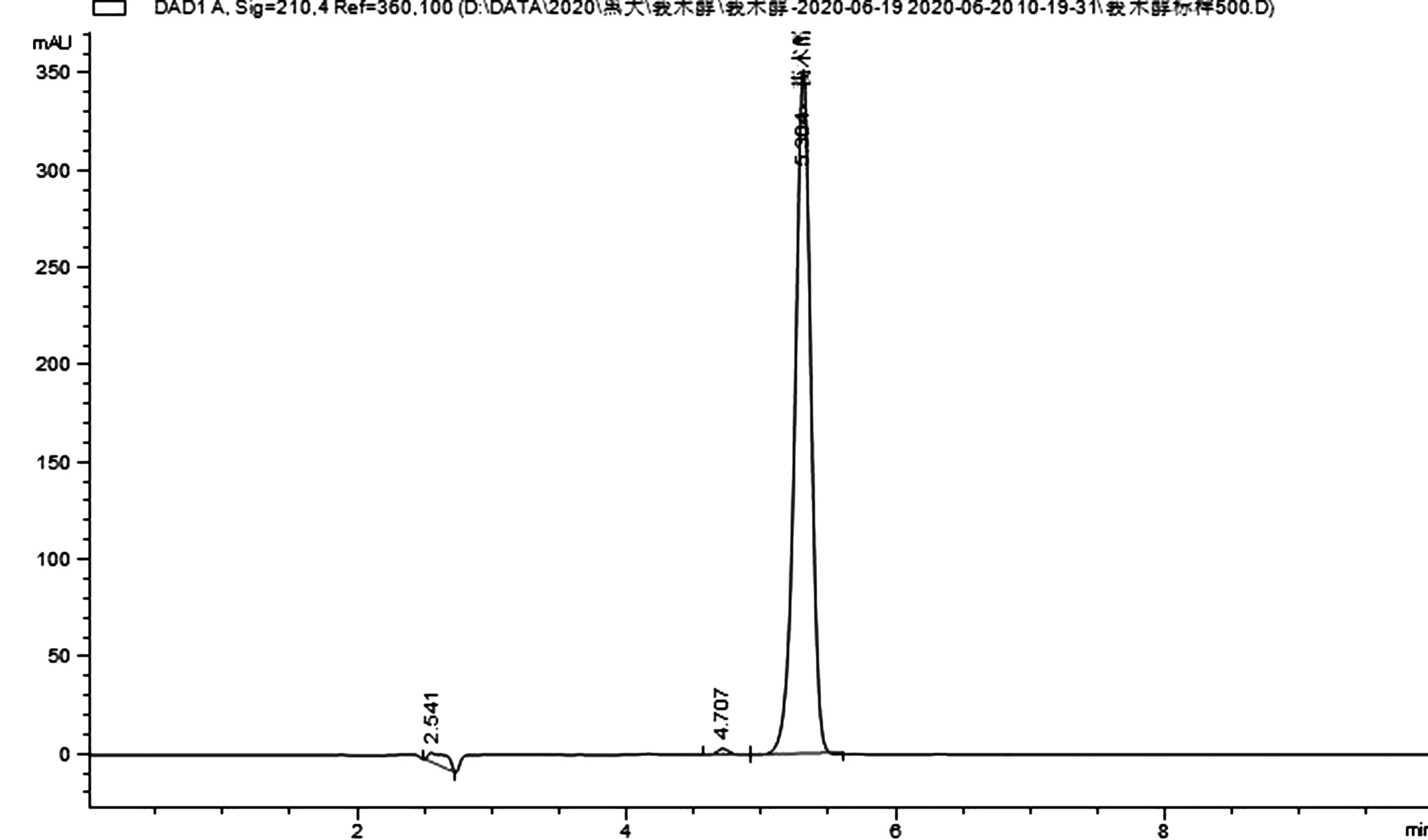
图1 莪术醇标品的液相色谱图
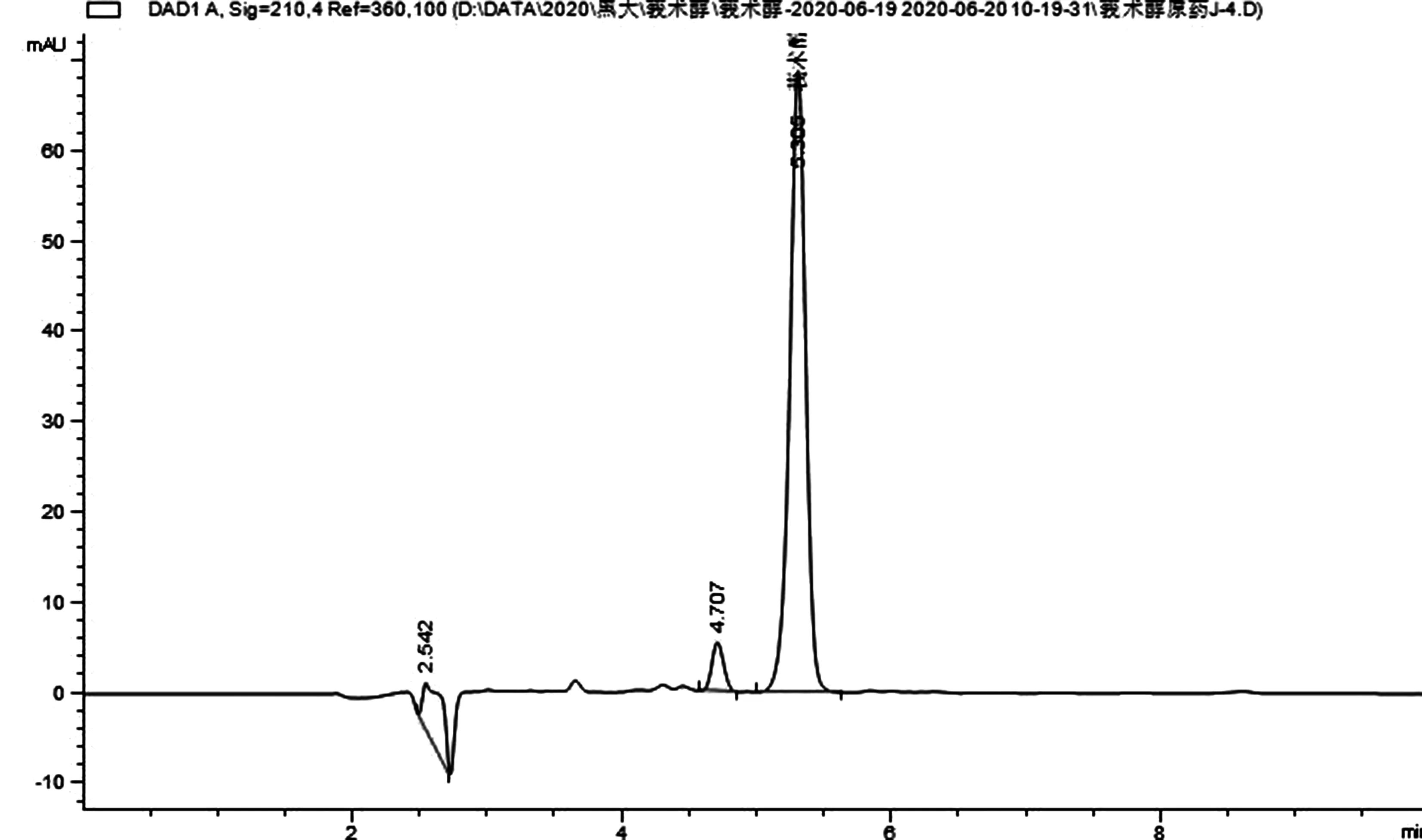
图2 莪术醇92%原药样品的液相色谱图
2.4.2 试样溶液的配制 称取试样0.026g(精确至0.2mg),置于100mL容量瓶中,用乙腈溶液超声溶解并稀释至刻度,静置备用。
2.4.3 测定 在上述操作条件下,待仪器基线稳定后,连续注入数针标样溶液,待各针相对响应值变化﹤1.5%时,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进样测定。
2.4.4 计算 将测得的2针试样溶液及试样溶液前后2针标样溶液中莪术醇的峰面积值分别进行平均,按下式计算其质量分数:
式中:
A1—标样溶液中莪术醇峰面积的平均值;
A2—试样溶液中莪术醇峰面积的平均值;
m1—标样的质量,g;
m2—试样的质量,g;
P —标样中莪术醇的质量分数,%。
3 结果分析
3.1 色谱条件的选择 通过Agilent ZORBAX 高效液相色谱仪的光谱数据采集功能,获得莪术醇的紫外吸收图(图3),从图中可以看到在吸收波长190~400nm区域内,莪术醇的最大吸收波长在200nm附近,由于吸收波长200nm附近溶剂干扰比较明显,因此将检测波长定为210nm。
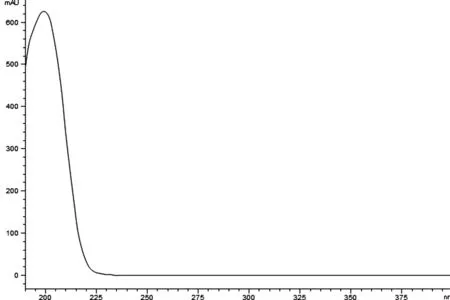
图3 莪术醇紫外吸收谱图
依据莪术醇的理化性质,选用乙腈和甲酸水溶液作为流动相,用乙腈溶解样品,有效成分与杂质能得到很好分离,峰形对称,基线平稳,分离效果好。
3.2 分析方法的线性相关性试验 准确称取莪术醇标样0.033g(精确至0.2mg),置于50mL容量瓶中,用乙腈超声溶解并稀释至刻度,静止备用。用移液管准确移取上述标准溶液2.0、4.0、6.0、8.0、10.0mL于10mL容量瓶中,用乙腈定容至刻度摇匀后备用。按上述条件进行测定,以进样浓度为横坐标,峰面积为纵坐标,绘制标准曲线,得线性方程为y莪术醇=9 632.27x-5.39,相关系数为0.999 9。
3.3 方法精密度的测定 在上述色谱操作条件下对同一样品平行测定5次莪术醇的标准偏差为0.10;变异系数为0.11%(表1)。

表1 分析方法的精密度试验结果
3.4 方法准确度的测定 按照配方比例要求,将除原药以外的所有填料助剂混匀,作制剂空白,按照制剂标称值在制剂空白中添加有效成分,合成5个制剂样品,在上述色谱操作条件下进行分析,测得莪术醇的平均加标回收率为99.93%(表2)。
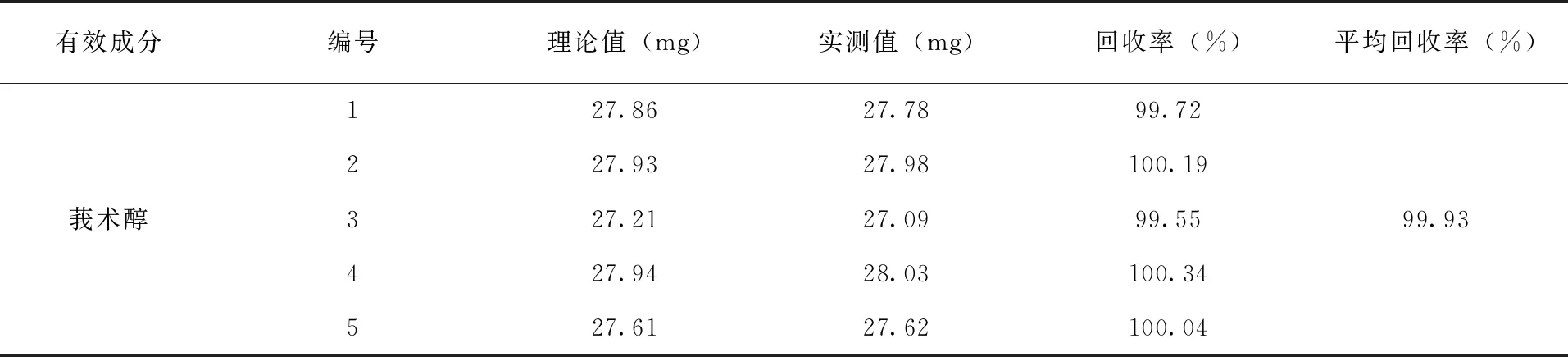
表2 分析方法的准确度试验结果
3.5 方法特异性 对样品组分色谱峰进行峰纯度试验,通过工作站软件计算匹配度,峰纯度因子为999.89,特异性符合要求。
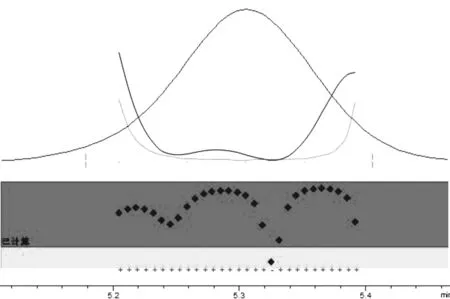
图4 莪术醇纯度
4 结论
本文建立了检测莪术醇原药中有效成分质量分数的高效液相色谱分析方法。试验结果表明,莪术醇在测试浓度范围内线性关系良好,有较高的准确度和精密度,具有简便、快速、分离效果好的优点,是一种可行的分析方法。
猜你喜欢
长春中医药大学学报(2022年6期)2022-11-23
广州化工(2022年18期)2022-10-23
云南农业科技(2022年1期)2022-01-27
中国应急管理科学(2021年4期)2021-04-13
中学化学(2019年4期)2019-08-06
考试周刊(2018年68期)2018-09-17
中国绿色画报(2018年4期)2018-08-09
农业与技术(2018年2期)2018-03-09
今日农药(2017年7期)2017-08-09
今日农药(2014年12期)2015-03-30
