
钨酸铋/石墨相氮化碳2D/2D光催化剂的制备及降解四环素的研究*
2021-04-11 08:27孙毓旋杨涵钰童睿真王珅韩松
化学与粘合 2021年2期
孙毓旋,杨涵钰,童睿真,王珅,韩松
(东北林业大学 林学院,黑龙江 哈尔滨150040)
引言
四环素类药物在畜牧业和人类医疗方面应用日益增加,此类污染物若直接排入水体当中,则会导致水体环境被大量污染;同时,也会沉积在土壤中渗透到地下水或经雨水冲刷直接排入水体。四环素类物质很难完全降解,现有的工艺只能部分去除四环素类抗生素,在降解过程中产生的中间产物很可能具有更大的毒性。一旦这些没有完全降解的中间产物被人体吸收后,将会对人体产生极大的危害性和抗药性。因此,在这一背景条件下,完全、高效地降解四环素类废水是当今亟待解决的一大问题。
当前,对于四环素类物质的降解研究主要集中在生物法、物化法、高级氧化法等,值得注意的是,上述方法均不能高效完全地对污染物进行降解,其降解效率仍然有待提升。综上所述,开发一种成本低、降解能力强、且绿色环保无污染的方法来解决此类问题尤为重要(对环境)。众所周知,光催化剂就具有上述优异性能,例如二氧化钛可以吸收太阳光中的紫外光,产生强氧化还原活性的电子和空穴对,从而将难去除的有机物转化为二氧化碳和水。但是二氧化钛禁带宽度为3.7V,仅能吸收太阳光中的紫外光,无法利用太阳光中占50%左右的可见光,并且电子和空穴极易复合,多余的能量以荧光的形式释放出来,影响其氧化还原能力。近年来,随着半导体技术的深入研究和开发,大量窄带隙的、具有可见光光催化活性的半导体被发现,例如:钨酸铋、类石墨相氮化碳等。它们的禁带宽度为2.7V,能够吸收部分可见光,具有良好的可见光光催化活性。但是,其电子空穴极易复合的这一特性,极大地削弱了其氧化还原能力,为了改正上述缺点,将两种或两种以上的催化剂进行复合,构建异质结,进一步调整其禁带宽度、能带结构,使光生电子和空穴进行有效的分离的同时,调整到合适的价带导带的位置形成适宜的氧化还原电势,从而对特定污染物进行降解。
本文用Bi2WO6/g-C3N4构建异质结,通过调节不同的负载量、pH值、反应温度等确定了异质结光催化剂的最优比例,此异质结具有合适的可见光吸收带隙宽度,针对盐酸四环素而言,具有合适的氧化还原电势,此异质结光催化剂在可见光下对盐酸四环素的降解率最高可达96.7%。
1 实验部分
1.1 主要原料与试剂
五水合硝酸铋,分析纯,上海麦克林有限公司;二水合钨酸钠,分析纯,上海麦克林有限公司;无水乙醇,分析纯,天津市密欧化学科技有限公司;盐酸四环素,分析纯,上海麦克林有限公司。
1.2 仪器与设备

表1 仪器与设备Table 1 The apparatus and equipment
1.3 实验步骤
1.3.1 热缩聚法制备g-C3N4
称取一定质量的二腈二胺放入坩埚中,将坩埚放入马弗炉中,以5℃/min的速率升温至550℃保持2h,冷却至室温,得到体相石墨相氮化碳,将其充分研磨,以备下一步使用。
1.3.2 水热法制备Bi2WO6/g-C3N4
将一定量五水合硝酸铋溶于20mL冰乙酸中搅拌20min,再加入30mL水充分搅拌30min,记为溶液A。将一定量二水合钨酸钠溶于20mL去离子水中超声30min,再加入一定量g-C3N4,超声1h,记为溶液B。将溶液B缓慢滴加到溶液A中,搅拌1h,用NaOH溶液调节溶液pH值。随后,将混合溶液转移至100mL高压反应釜中,160℃分别反应8h、12h、16h,冷却至室温后将反应釜取出,离心取出产物后用去离子水和无水乙醇交替洗涤数次,将产物于80℃烘干,取出研磨成粉末,即得到Bi2WO6/g-C3N4异质结。Bi2WO6/g-C3N4的质量比为40%、60%、80%,pH值分别调节至3、5、7、9,反应时间分别为8h、12h、16h。
1.4 Bi2WO6/g-C3N4异质结光催化降解盐酸四环素性能的测试
光催化实验装置由磁力搅拌器、光催化反应器、光源和遮光罩组成。其中以350W氙灯为催化光源,用来模拟太阳光照射,由于紫外光仅占太阳光的5%,为了模拟最真实太阳光光源,我们并不考虑加紫外滤光片。量取100mL,40mg/L的盐酸四环素加入到光催化反应器中,将光催化反应器放在磁力搅拌器上进行匀速搅拌,加入0.1g Bi2WO6/g-C3N4,盖上遮光罩,进行暗反应30min,以测量催化剂的吸附平衡。在氙灯下进行光催化反应,用注射器每30min取一次样,将液体通过0.45vm的微孔滤膜注射到比色皿中,运用紫外分光光度计测量吸光度。根据朗博-比尔定律,溶液的吸光度与吸光物质的浓度呈正比,所以盐酸四环素的降解率可以用吸光度来表示,光催化效果用盐酸四环素的降解率表示:
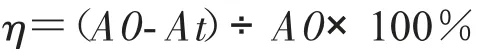
其中A0为盐酸四环素初始吸光度,At为盐酸四环素降解后的吸光度。
2 结果与讨论
2.1 X射线衍射表征(XRD)
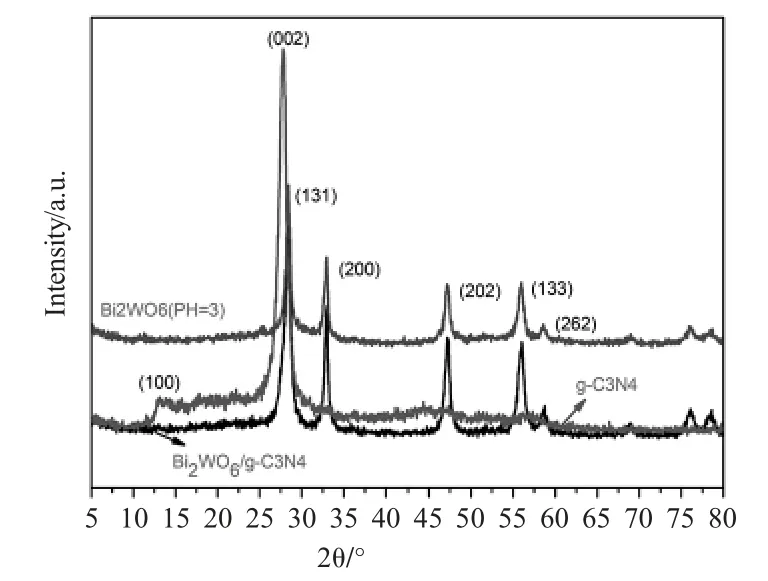
图1 Bi2WO6 XRD、g-C3N4 XRD、Bi2WO6/g-C3N4的XRDFig.1 The XRD pattern of Bi2WO6,g-C3N4 XRD and Bi2WO6/g-C3N4
如图1所示,本文使用了X射线衍射技术探究纯相Bi2WO6、纯相g-C3N4和g-C3N4/Bi2WO6的元素组成[4]。g-C3N4/Bi2WO6的特征衍射峰分别与类石墨相g-C3N4标准卡片No.87-1526[4]、斜方晶系Bi2WO6的标准卡片No.73-1126相匹配。在28.3°,32.8°,47.1°,56.0°和58.5°处发现了样品的明显衍射峰,分别对应于(131),(200),(202),(133)和(262)晶面[1]。对于纯的g-C3N4而言其(100)晶面和(200)[2]晶面分别对应2θ=13.6°和27.4°,2θ=27.4°是由共轭芳香环体系典型的层间堆积造成的。g-C3N4和Bi2WO6的主峰在27.4°处重叠,因此无法检测到样品中g-C3N4的特征峰,没有发现其它杂质。
2.2 透射电镜测试(TEM)

图2 (a)、(b):g-C3N4的TEM;(c):g-C3N4的HRTEM;(d)、(e):Bi2WO6/g-C3N4的TEM;(f):Bi2WO6/g-C3N4的HRTEMFig.2(a),(b):The TEM images of g-C3N4;(c):The HRTEM image of g-C3N4;(d),(e):The TEM images of Bi2WO6/g-C3N4;(f):The HRTEM image of Bi2WO6/g-C3N4
我们通过HRTEM进一步研究了g-C3N4和Bi2WO6/g-C3N4的微观形貌。如图2(a)和(b)所示,纯的g-C3N4有大小不一的薄片堆叠形成的薄层状纳米片,仅有小部分纳米片层数较多;如图2(c)所示,大部分g-C3N4是体相高度剥离二维薄层状态,其(002)面的结晶度也在减小,故HRTEM无法看到晶格间距。仅有一小块区域可看到晶格间距,其晶格间距为0.32nm,对应的是g-C3N4的(002)面,根据HRTEM分析,初步断定这种情况形成的原因为剥离不够彻底所引起的多层体相g-C3N4残留,其层数较多,结晶度较好。如图2(d,e)所示,Bi2WO6/g-C3N4在不同倍率下TEM均体现出纳米片的结构,g-C3N4为载体,Bi2WO6紧密连接在g-C3N4上,该异质结由两种不同的二维纳米薄片自组装形成。从图2(f)中可以直观地看出Bi2WO6和g-C3N4的交界面,由于薄层状g-C3N4层数较少所以结晶度不高,与之形成明显对比的是Bi2WO6/g-C3N4的异质结,可以明显看出0.27nm的晶格间距,其对应的是Bi2WO6的(200)面[5]。综上所述,Bi2WO6/g-C3N4的异质结初步形成,接下来通过XPS进一步进行分析论证。
2.3 X射线光子能谱(XPS)
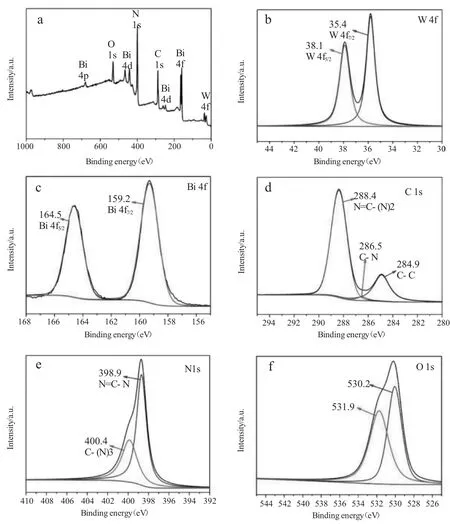
图3 Bi2WO6/g-C3N4的XPS能谱;(a)全谱;(b)W 4f;(c)Bi 4f;(d)C 1s;(e)N 1s;(f)O 1sFig.3 The XPS energy spectrum of Bi2WO6/g-C3N4;(a)Full spectrum;(b)W 4f;(c)Bi 4f;(d)C 1s;(e)N 1s;(f)O 1s
为了进一步确定Bi2WO6/g-C3N4的表面元素组成和化学价态,我们研究了Bi2WO6/g-C3N4的XPS。从Bi2WO6/g-C3N4异质结的XPS全谱可以明显看到Bi、O、N、W、C元素的峰。在高分辨率的Bi 4f的谱图中,159.2eV和164.5eV分别对应于Bi 4f7/2和Bi 4f5/2这两个峰[4]。在35.4eV和38.1eV的两个峰分别对应于W 4f7/2和W 4f5/2。C 1s谱图中284.9eV、286.5eV、288.4eV的结合能分别对应C-C、C-N和N=C-(N)2基团[4]。如图所示,530.2eV和531.9eV的结合能分别代表Bi2WO6晶格中的氧和Bi2WO6中与外界成键的氧。Bi2WO6中的氧与g-C3N4中C1s轨道的C-C键、C-N键和N=C-(N)2键中的C成键,因为在C 1s谱图中可以看出的C 1s轨道C的结合能相比于标准物质均增大,表明C在异质结的形成过程中失去电子,而在O 1s的峰中,531.8eV的结合能代表与g-C3N4成键的O,因此可以证明异质结形成[7]。如图所示,在N 1s谱图中398.9eV和400.4eV的结合能分别对应N=C-N和C-(N)3基团。综上所述,XPS表明了C-O键的存在,进一步证明了Bi2WO6/g-C3N4异质结的形成。
2.3 样品的紫外-可见光漫反射分析
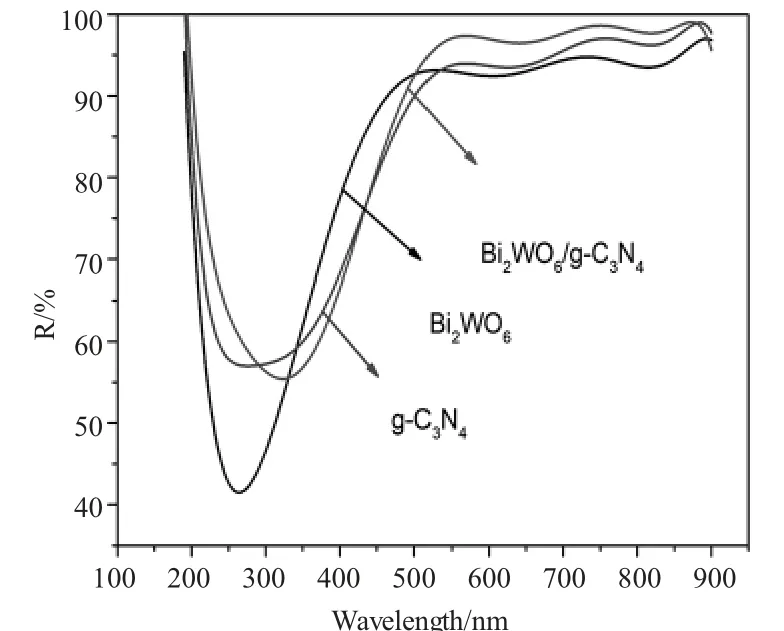
图4 Bi2WO6/g-C3N4、Bi2WO6、g-C3N4的紫外-可见光漫反射Fig.4 The ultraviolet-visible diffuse reflection of Bi2WO6/g-C3N4,Bi2WO6,g-C3N4
本实验通过紫外-可见漫反射表征了样品的光吸收性能,如图4所示,钨酸铋的可见光吸收波长最窄,Bi2WO6/g-C3N4的可见光吸收波长最宽,g-C3N4在二者之间,可以说明以g-C3N4为载体的Bi2WO6/g-C3N4异质结拓展了Bi2WO6的可见光吸收范围。
2.4 光催化性能测试
2.4.1 不同Bi2WO6/g-C3N4的质量比对光催化性能的影响
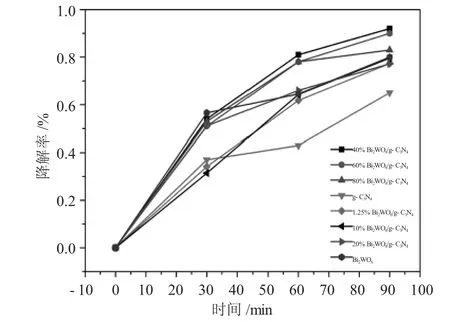
图5 不同负载量光催化降解图Fig.5 The photocatalytic degradation with the catalyst with different loads
如图5所示,纯相的钨酸铋光催化降解盐酸四环素效果要好于纯相的类石墨相氮化碳,钨酸铋和石墨相氮化碳的降解率分别为83.7%和77.2%,并且从图中可以看到前30min纯相钨酸铋的降解效果最好,后60min降解率趋于平缓,可能是因为钨酸铋的光生电子和空穴容易复合,降低了钨酸铋的使用寿命。本实验分别尝试了不同质量比为1.25%
Bi2WO6/g-C3N4、10%Bi2WO6/g-C3N4、20%Bi2WO6/g-C3N4、40%Bi2WO6/g-C3N4、60 %Bi2WO6/g-C3N4、80%Bi2WO6/g-C3N4的光催化降解盐酸四环素效果,如图所示,当负载量为40%Bi2WO6/g-C3N4时降解效果最好,其降解率为92.2%;当负载量低于40%时催化效果随着负载量的降低而下降,当负载量下降为1.25%Bi2WO6/g-C3N4时降解效果最差,其降解率为75.5%。当负载量由40%增加至80%的过程中,催化剂的催化效果也随着负载量的增加而下降,其中80%Bi2WO6/g-C3N4的降解效率为79.7%,略好于最差负载量1.25%Bi2WO6/g-C3N4的降解效果。综上所述,当其他条件一定时,负载量为40%Bi2WO6/g-C3N4的降解效果最好。
2.4.2 合成催化剂时不同pH值光催化降解盐酸四环素的降解效率
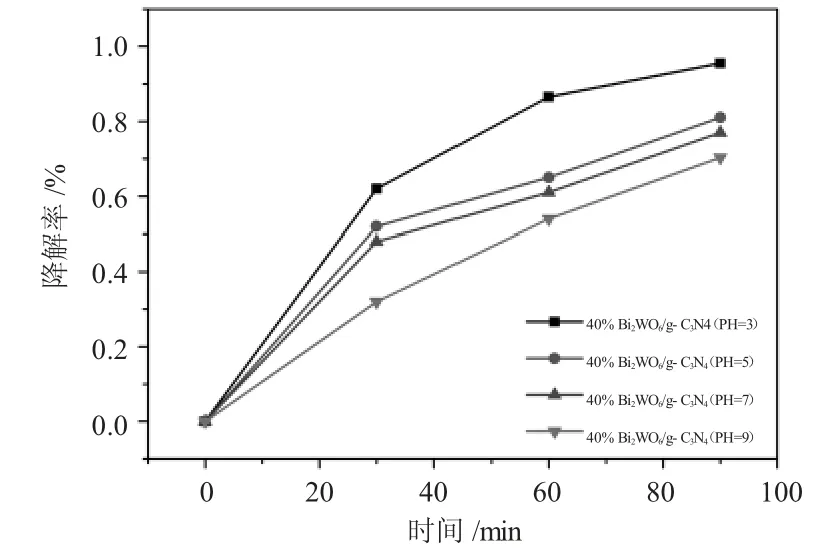
图6 不同pH值光催化降解图Fig.6 The photocatalytic degradation under the condition of different pH values
如图6所示,当负载量均为40%Bi2WO6/g-C3N4,合成时间均为12h时,催化剂降解盐酸四环素的效率随着pH值的增加而明显下降,当pH值等于3时降解效果最好,其降解效率为95.5%;当pH值等于9呈碱性时,催化剂的降解效果最差,其降解效率为70.3%。造成这种现象的原因大概是硝酸铋水溶液为强酸弱碱盐,其在水溶液中极易水解,所以保证溶液中足够的氢离子浓度能有效地抑制其水解;钨酸钠的水溶液为强碱弱酸盐,其极易与氢离子结合生成白色的钨酸沉淀,生成的钨酸沉淀成为了钨酸铋成核的关键位点,当氢离子浓度过高生成钨酸数量过多时,成核位点较多而铋离子水解缓慢,不利于反应的进行;当pH值过高时,成核位点过于少,硝酸铋水解过快,不利于纳米片的生长,综上所述,pH值对催化剂的光催化性能影响至关重要。
2.4.3 不同合成时间的催化剂光催化降解效率
如图7所示,当反应负载量为40%Bi2WO6/g-C3N4、合成的pH值为3时,随着催化剂合成时间的增加,该催化剂光催化降解盐酸四环素的效率先升高后下降,当反应时间为12h时降解效率最高,其降解效率为96.7%;当反应时间为16h时,该催化剂光催化降解盐酸四环素的效率最低,其降解效率为77.8%。引起此现象的原因为:所有晶体都要经历成核和生长的过程,反应初期大部分核生长完成,反应时间越长晶体生长得越大,经过不断地生长堆积,最终形成比较厚大的纳米片,影响了催化剂的催化效果。

图7 催化剂不同合成时间光催化降解图Fig.7 The photocatalytic degradation with the catalysts prepared with different synthesis time
3 结论
本文采用水热法合成Bi2WO6/g-C3N4异质结光催化剂,经过实验证明当异质结负载量为40%Bi2WO6/g-C3N4、水热时间为12h、pH值为3时其光催化剂降解盐酸四环素效果最好,降解效率高达96.7%。为了证明Bi2WO6/g-C3N4异质结光催化剂催化效果很强,本实验做了纯相钨酸铋和类石墨相氮化碳进行对比,Bi2WO6/g-C3N4异质结光催化剂的光催化降解盐酸四环素的效率是纯相钨酸铋的1.2倍、是纯相类石墨相氮化碳的1.3倍。本文通过XRD证明了钨酸铋、类石墨相氮化碳晶型的形成,初步确定了异质结的形成。通过HRTEM表征手段,可直观地看出超薄g-C3N4的形成和Bi2WO6/g-C3N4异质结的形成,同时通过晶格间距的测量进一步证明了XRD表征的合理性。XPS能谱分析确切证明了C 1s轨道C成键的结合能的增加和Bi2WO6中与C成键O的存在。通过这几种表征手段确切证明了Bi2WO6/g-C3N4异质结的形成,并且通过光催化降解实验证明了Bi2WO6/g-C3N4异质结光催化剂对难降解抗生素盐酸四环素的降解效果的高效性。
猜你喜欢
湘潭大学自然科学学报(2022年2期)2022-07-28
石油化工高等学校学报(2022年1期)2022-04-15
陶瓷学报(2021年5期)2021-11-22
烟台果树(2021年3期)2021-07-21
陶瓷学报(2021年1期)2021-04-13
粉末冶金技术(2021年1期)2021-03-29
陶瓷学报(2020年6期)2021-01-26
陶瓷学报(2020年3期)2020-10-27
中外葡萄与葡萄酒(2019年2期)2019-04-12
热处理技术与装备(2019年1期)2019-03-14
