
固体在液相中吸附热力学参数计算介绍
2021-04-09 11:15:54王伟涛陈香李杨百勤
大学化学 2021年2期
王伟涛,陈香李,杨百勤
陕西科技大学化学与化工学院,西安 710021
吸附现象在实际生活和生产中广泛存在。吸附是指在固相-气相、固相-液相、液相-液相等体系中,气相或者液相组分在表面上发生富集的现象[1]。固体在液相中吸附应用十分广泛,如染色、脱色、废水处理、水净化、液相组分的分离和提纯等方面。
固体表面能够发生吸附现象主要由于固体表面的不均匀性。固体表面并不是简单的体相中止,也不是体相结构的简单延续。从原子水平看,固体的表面并不是光滑的,表面的原子存在很多种可能的结构环境(图1)。所以,固体表面原子处于化学键不饱和状态,具有较高的能量,即热力学上处于不稳定状态,必然会自发地吸附气体或溶液中的分子,降低其表面自由能。因此,吸附过程是自发的自由能减小过程。

图1 固体表面原子情况示意图[2]
被吸附的物质称为吸附质(adsorbate),具有吸附作用的物质称为吸附剂(adsorbent)。吸附作用力有化学键、氢键、酸碱、共轭π-π作用、静电力、范德华力等作用力。吸附热力学参数可以判断吸附过程能否自发进行,吸附自发进行的热力学趋势大小;通过吸附热力学可以进一步分析吸附质、吸附剂、溶剂等吸附体系和条件对吸附过程的影响[3]。用热力学方法处理吸附平衡问题时,一般需要热力学参数来对其进行分析。因此,研究固-液吸附时需要以吸附热力学参数来分析其吸附热力学,即吸附吉布斯自由能变(ΔG)、吸附焓变(ΔH)、吸附熵变(ΔS)。
尽管吸附热力学已经成为研究吸附现象的必要手段,但是没有专门的文献介绍吸附过程热力学参数的计算方法,致使初学者在研究吸附热力学性质时需要查阅大量的文献。另外,在众多文献报道中吸附热力学参数计算方法不同,并且文献中一般只是直接引用所使用的计算公式,这对初学者来说会带来很大的困惑。因此,本文通过介绍不同方法计算吸附热力学参数的推导,并通过举例介绍吸附热力学参数的计算和应用。
1 固-液界面吸附过程化学势
固-液界面的吸附可以看作特殊的相变过程,吸附平衡是一种特殊形式的相平衡[4,5]。因此,可以利用相平衡原理来处理吸附平衡。液相中某组分i在固-液界面上发生吸附,吸附过程是组分i从液相中迁移到固-液界面上:

达到吸附平衡时,根据组分i在液相中和界面上的化学势相等,可以建立关系式。
液相中组分i的化学势:

吸附在固体表面的组分i化学势:
式中,fi和fads,i分别为组分i在液相和在吸附相中的活度因子;ci和cads,i分为组分i在液相和吸附相中的浓度;ai和aads,i分为组分i在液相和吸附相中的活度。



2 固-液界面吸附过程ΔG计算
从式(7)中可以看出,要计算吉布斯函数变化ΔGө,需要先计算吸附平衡常数K。K值是计算吸附过程热力学函数变化的关键,但是不同文献中对K的计算方法各不相同。因此,我们有必要厘清K的计算。
2.1 极限法获得K计算ΔG

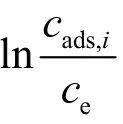

式中,ρ1为溶剂的密度(g·mL−1),M1为溶剂的分子量(g·mol−1),A1为溶剂分子的横截面积(cm2·molecule−1),s为吸附剂的表面积(cm2·g−1),x/m 为比吸附量(μg·g−1),M2为吸附的组分 i分子量(g·mol−1),式中得到 cads,i的单位为 mol·L−1。这样就可以求出 K。

2.2 利用平衡浓度获得K计算ΔG
直接使用溶液中溶质的浓度变化量和平衡浓度的比值计算K[11],即

式中,c0为溶液中溶质的初始浓度,ce为平衡浓度。利用此式可以简单迅速地计算出K。该式是以(c0− ce)代替了吸附相中组分i的浓度cads,i;只有在稀溶液下才可以近似认为活度因子为1,和式(8)一致,因为K的定义为活度之比,否则也需要利用极限法计算。在溶液中吸附,(c0− ce)和cads,i存在一定的差异;当吸附剂对溶剂的吸附量大于对溶质的吸附时,此时(c0− ce)为负值,从而导致K为负值,而无法利用式(7)计算ΔG;而实际上,cads,i只可能为正值。因而,该方法计算并不是很准确,仅仅可以粗略估算吸附热力学参数的变化趋势。
直接使用平衡吸附量和平衡浓度的比值计算K[12],即

式中,qe为吸附平衡时吸附剂对吸附质的表观吸附量(mol·g−1),ce为平衡浓度(mol·L−1)。该式是在式(10)的基础上,认为在极低稀溶液时,在吸附相上吸附量非常小,组分i在吸附相的浓度数值上约等于吸附量。因而,该式仅限于无限稀溶液下的计算;同时,也存在式(10)的缺陷:qe和K有可能也为负值,而无法计算ΔG。另外,式(11)计算存在的另一个问题是K有量纲(L·g−1)。为此,有研究者提出[3],K值应该乘以溶剂的浓度(g·L−1),即可得到较为合适的K。
2.3 利用等温吸附模型中的常数计算
固体在液相中的吸附体系远比固-气界面的吸附复杂。固体在液相吸附时,固体表面存在对溶质和溶剂的竞争吸附,而这种竞争是溶质-溶质、溶剂-溶剂、溶剂-溶质、溶质-吸附剂、溶剂-吸附剂之间作用的综合结果。目前,液相吸附的理论多是沿用气体吸附的理论模型,虽然液相吸附的实验结果在形式上能用气体吸附模型处理,但其也只是经验性的。
根据式(7),只要求出吸附过程的平衡常数K,就可以求出 ΔG。通过拟合等温吸附过程的吸附模型,直接以等温吸附模型方程式中的吸附常数代替K来计算ΔG[13]。常见的等温吸附模型如表1所示。文献中报道最多的是Langmuir和Freundlich方程,多数吸附等温模型符合这两个模型中的一个。当符合Langmuir方程或Freundlich方程时,直接用方程中的KL或KF代替K[14]计算ΔG。当符合Herry方程,以Herry方程中的常数KH计算时,有KH=qe/ce,与式(11)一致。利用等温吸附模型中的常数计算吸附热力学参数,可以同时获得符合的等温吸附模型和吸热的热力学参数。需要指出的是,不同吸附模型得到的ΔG之间不具有可比性,毕竟K和等温模型方程式中的吸附常数不同;但是得到的计算结果并不影响其吸附热力学上的趋势规律。

表1 不同的吸附等温模型及其方程表达式
2.4 利用Freundlich公式参数法计算ΔG
Gibbs吸附等温式可以写成[15]:

式中,ΔGʹ为吸附自由能变(J),a为溶质在溶液中的活度,N为吸附的溶质摩尔数(mol)。为了获得单位质量的吸附吉布斯自由能变化,式(13)可以变化为:

式中,ΔGʹ为吸附自由能变(J·g−1),q为吸附量(mol·g−1)。当浓度较低时,可以用浓度或摩尔分数代替活度[15]。用摩尔分数代替活度时,式(14)变为:

式中,x为溶液中溶质的摩尔分数。将Freundlich方程式中的吸附量q = KFx1/n带入式(15)中,可以得到:

积分后,得

对ΔG′′除以q,可以得到每摩尔溶质吸附的吉布斯自由能变(J·mol-1),即

这是一个和吸附量无关的吉布斯自由能变的计算公式,式中n为Freundlich方程式中的常数,T为温度(K)。一般来说,对于符合Freundlich吸附模型的等温吸附来说,在计算Gibbs自由能变时,只要获得Freundlich吸附模型方程式中的常数n,就可以直接用ΔG = −nRT即可。
在研究吸附过程吉布斯自由能变时,需要根据实际情况选择合适的计算方法。采用不同方法计算的吸附自由能变数值可能不同,但是其变化规律是一致的。因此,如果要比较不同吸附体系的热力学函数ΔG,需要采用同一种计算方法。一般来说,物理吸附的自由能变化绝对值小于化学自由能变绝对值,物理吸附自由能变一般为−20 - 0 kJ·mol−1,化学吸附的自由能变一般为−400 - −80 kJ·mol−1。因此,可以通过ΔG的数值来判断吸附过程是物理吸附还是化学吸附过程。
3 固-液界面吸附过程ΔH计算
3.1 利用平衡常数K求ΔH
吸附焓变(或者吸附热)可以通过Van’t Hoff方程计算[16]:

式中,K为吸附平衡常数。设ΔH不是温度的函数,可以对其进行积分,得到:

通过不同的温度下的平衡常数K和温度T,可以求出吸附焓变ΔH。式(21)也可以由ΔG = ΔH −TΔS直接得到。将ΔG = −RTlnK带入,可得到:

由式(23)可以知道,式(21)中的常数C为ΔS/R。利用式(21)或者式(23),以不同温度下的lnK对1/T作图,得到一条直线,根据直线斜率求出ΔH。文献也有此式的变化形式[17]:

式(21)、式(23)、式(24)本质上是一致的。
3.2 利用平衡浓度ce求ΔH
在稀溶液中,活度可以近似等于摩尔分数;同时,假定在吸附量一定时,吸附剂上吸附的溶质和溶剂的比例是常数;可以得到平衡常数和平衡时溶液中溶质摩尔分数成反比,和平衡时溶液中溶质的浓度成反比[15],即:

将其带入Van’t Hoff方程的定积分式(120)中,则Van’t Hoff方程变为:

式中,ce,1和ce,2分别表示不同温度下达到吸附平衡时的浓度。
将式(25)带入Van’t Hoff方程的不定积分式(21)中,可以得到:

通过lnce对1/T作图,得到一条直线,根据直线的斜率可以求出ΔH。该式和文献中报道的利用Clausius-Clapeyron方程求解ΔH的方程式形式一致[18]。
除此之外,ΔH还可以利用Gibbs-Helmholtz方程计算[10]:

只要求出了ΔG,可以方便的求出ΔH。
通过吸附热的大小可以粗略判断吸附作用力类型[19,20]。吸附作用力有化学键、氢键、电子供体受体作用、共轭π-π作用、静电力、范德华力等作用力。当吸附作用力为范德华力时,吸附热在4-10 kJ·mol−1;吸附作用力为氢键时,吸附热为 2-40 kJ·mol−1;作用力为配位基交换、偶极间作用力时,吸附热分别为~40和2-29 kJ·mol−1;当吸附作用力为化学键时,吸附热大于60 kJ·mol−1。但是,吸附过程的吸附热是吸附质和吸附剂之间多种作用力的综合作用结果,通过吸附热判断作用力的类型只是一个粗略的估计。另外,可以用吸附焓变来区分吸附过程是化学吸附或物理吸附。一般认为,焓变的绝对值< 40 kJ·mol−1时,为物理吸附;焓变的绝对值在50-200 kJ·mol−1时,则为化学吸附。如果吸附焓变为负值,则表示吸附过程是放热的,升高温度将不利于吸附;反之如果吸附焓为正值,则表示吸附过程是吸热的,升高温度将利于吸附。
4 固-液界面吸附过程ΔS计算
得到ΔG后,ΔS可以根据下式进行计算[21]:

在求 ΔH时,可以利用式(23),以 lnK对1/T作图,利用直线的截距 ΔS/R可以计算出ΔS。另外,用已经求出的ΔG和ΔH,通过Gibbs-Helmholtz方程直接计算:

在固体自气相吸附,气体由三维的气相转移到二维表面相时,其自由度降低,气体的吸附过程伴随的是熵的减少,熵变为负值;所以,气相物理吸附的驱动力一般是焓变而不是熵变。但是在液相吸附时,溶质和溶剂都会发生吸附,溶质的吸附是熵减少的过程;溶质的吸附必然伴随溶剂的脱附,溶剂分子的脱附是熵增过程[10]。所以,吸附过程熵变应该是二者共同作用的结果。如果吸附过程仅仅是物理吸附,同时溶质分子面积大于溶剂分子面积时,意味着吸附一个溶质分子可能会有大于一个的溶剂分子脱附;所以,溶质吸附引起的熵减可能小于溶剂分子脱附引起的熵增,其吸附过程的熵变可能是正值。因此,吸附过程的熵是增大还是减小,和溶质和溶剂分子的相对大小有关。
5 应用举例
5.1 活性炭自水溶液中吸附对二甲苯
KC-8活性炭自水溶液中吸附对二甲苯,通过对其吸附等温线拟合,发现其符合Freundlich吸附模型,因此可以根据式(18)计算ΔG,根据式(27)计算ΔH,根据式(30)计算ΔS,得到的吸附热力学参数如表2所示[22]。该吸附过程的ΔG <0且其值在−20 - 0 kJ·mol−1范围内,表明该吸附过程是一个自发的物理吸附过程。该吸附过程的ΔH> 0,表明该吸附过程是吸热过程;根据ΔH值的大小并结合ΔG值可以判断,该吸附作用力不可能为化学键作用力。吸附熵变ΔS> 0,因此可以推断该吸附过程伴随着溶剂的脱附过程。当活性炭吸附对二甲苯时,其自由度减少,对应的熵减少;当溶剂分子(水)脱附时,熵值增大。当吸附一个对二甲苯分子时,就会有多个溶剂水分子脱附,因而该吸附过程总熵变大于0。

表2 在313.15 K下KC-8活性炭自水溶液中吸附对二甲苯的热力学参数[22]
5.2 硅胶对同系物的吸附
硅胶自四氯化碳中吸附脂肪醇同系物,计算得到的ΔG如表3所示[23]。从表中可以看出随着脂肪醇碳链的增长,对应的ΔG增大,表明在非极性溶剂中极性吸附剂对极性吸附质的作用较强。随着碳链长度增加,每一个CH2官能团对ΔG的贡献值并不是一个常数。这表明脂肪醇并不是以“平躺式”在界面吸附。在稀溶液中,单官能团极性有机物是以其极性基团吸附在硅胶表面,非极性的疏水基团以一定的方式留在非极性溶液中[5]。

表3 硅胶从正脂肪醇的四氯化碳溶液中吸附的ΔG [23]
硅胶从环己烷中吸附苯、萘、蒽、菲的热力学参数如表4所示[21]。硅胶对苯、萘、蒽、菲的吸附ΔG差别不大。这是由于这四种物质的结构相似,在硅胶表面的吸附都是由π电子对固体表面的作用而吸附,吸附时都是“平躺”在界面上的。

表4 硅胶自环己烷中吸附苯、萘、蒽、菲的热力学参数[5]
6 结语
吸附过程的热力学参数对研究吸附剂吸附性质具有重要的指导意义。吸附热力学参数的计算方法有多种,不同的计算方法得到计算结果可能不同。因此,不同计算方法得到计算结果不具有可比性;但是不同计算方法不改变吸附热力学参数的变化规律。通过获得的热力学参数可以判断吸附过程是物理吸附或化学吸附,也可以判断吸附质和吸附剂之间作用强弱及粗略判断吸附作用力的类型;同时,可以通过吸附的热力学参数之间的比较可以推断溶质分子结构对吸附的影响及吸附方式。
猜你喜欢
初中生学习指导·中考版(2024年1期)2024-04-19 09:30:00
阅读(科学探秘)(2022年8期)2022-05-30 10:48:04
初中生学习指导·中考版(2022年1期)2022-02-09 22:34:56
中学生数理化·中考版(2017年12期)2017-04-18 12:55:09
上海金属(2016年1期)2016-11-23 05:17:24
中学生数理化·高三版(2016年2期)2016-09-10 07:22:44
电子制作(2016年19期)2016-08-24 07:49:54
山西大同大学学报(自然科学版)(2015年1期)2015-01-22 07:14:08
中国卫生(2014年5期)2014-11-10 02:11:32
机械与电子(2014年2期)2014-02-28 02:07:43
