
催化剂相关概念的辨析
2021-04-09 11:15:52凌晨张树永
大学化学 2021年2期
凌晨,张树永
山东大学化学与化工学院,济南 250100
人类使用酵母和酒曲制作酒、醋的历史可以上溯到史前。近代的催化作用研究始于 18世纪后期,最早的报导是1781年Pamentire发现酸也可以促进淀粉的糖化;1792年,Scheele发现酸可以促进酯化反应,而碱可以促进皂化反应;1794年,Fulhame在其书中首次提出催化的概念。1835年,瑞典人Berzelius给出了催化作用的最早定义:
“催化反应是可以被反应后性质不变的物质加速的反应。”
期间,1795年,Deiman利用炽热的黏土使乙醇脱水变成乙烯是第一多相催化的实例。1806年,Clement和Desormes对当时已经广泛采用的铅室法制硫酸进行了分析和改进。这些新现象、新体系的发现和研究,对催化和催化剂概念的形成和发展产生了推动作用[1-3]。
催化过程是非常重要的物理化学过程,催化化学已经发展成为相对独立的物理化学三级学科,并有向催化科学发展的趋势。在物理化学教科书和催化化学专著中,几乎都会对催化剂及催化作用进行分析和阐述,但由于着重点不同,导致相关表述千差万别。催化剂和催化作用的定义也是众说纷纭。1996年,IUPAC给出了催化作用和催化剂的新定义[4]:
A substance that increases the rate of a reaction without modifying the overall standard Gibbs energy change in the reaction; the process is called catalysis. The catalyst is both a reactant and product of the reaction. (能够增加反应速率而不改变反应总的标准吉布斯自由能变化的物质是催化剂。相关过程称为催化作用。催化剂既是反应的反应物也是产物。)
按照IUPAC的新定义,一个一般的催化反应的方程式应该写成式(1)的形式

其中A、B为反应物,G、H是产物,a、b、g、h是其计量系数。C表示催化剂。显然,该式与之前常用的表示催化反应的方程式(式(2))

将催化剂作为反应条件写在反应箭头上面的做法不同。
距离该定义的发表已经过去了近四分之一世纪,在此之后大量新出版和修订的教科书和专著均未采用该定义。这不能不说是一个问题。
本文对国内外主要教科书中的催化剂定义进行了对比分析,试图进一步明确催化剂的特征及其定义的要点,并给出建议。
1 催化剂定义的对比分析
要辨析如何定义催化剂和催化作用,我们必须首先明确催化剂的内涵及其本质特征。
1835年Berzelius指出,催化剂不参与化学反应[1],反应后不发生变化,但却可以改变反应。指明了催化剂不参与反应、性质不变、改变反应速率3个特征。随着人们对催化剂和催化作用研究的日渐广泛深入,人们对催化剂的本质特征有了更多的了解。概括而言,催化作用的特征主要包括:外加(addition)、物质(substance)、用量(amount)、速率(rate: change/accelerate/speed up 改变/加快/加速)、质量(mass)、化学性质(chemical nature)、参与反应(take part in the reaction)、反应途径(reaction route/pathway)、活化能(activation energy)、热力学特征如平衡(equilibrium)、平衡常数(equilibrium constant)、Gibbs自由能(Gibbs free energy)、转化率(conversion)、产率(yield)、催化剂是条件还是反应物和产物(conditions or reactant and product)、催化剂是否存在反应计量关系 (stoichiometry) 等[3,4,5-11]。表1针对这些特征,对重要物理化学教科书中的催化剂定义和特征进行了列表对比。
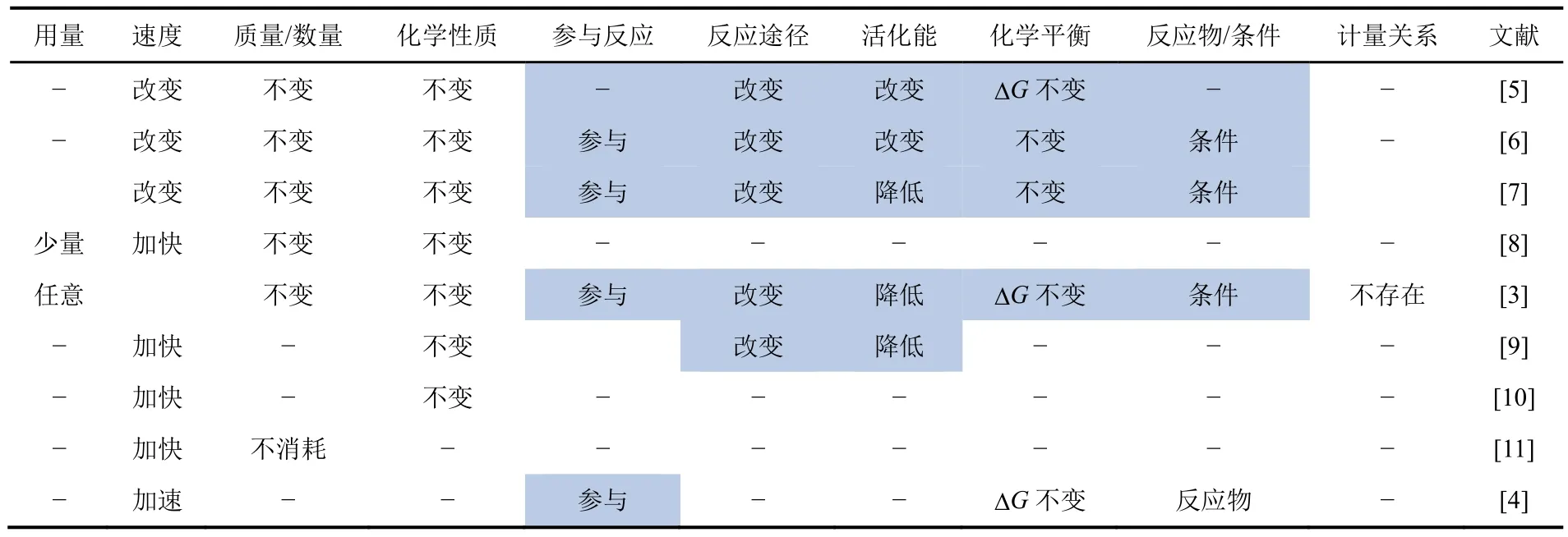
表1 主要教材和专著给出的催化剂定义和特征分析的比较
下面逐一对相关特征进行分析和说明。
(1) 催化剂是外加的。有不少教科书[6,7]、专著和文献[12]认为,催化剂不是体系固有的,需要人为外加。但实际上催化剂可以由反应体系生成,即存在自催化现象[3,6]。因此,外加不是催化剂的本质特征。
(2) 催化剂是物质。该要点的含义是催化剂是物质而非能量。催化剂可以是单质、合金、化合物、复合物、混合物甚至离子;可以是单一物质也可以由多种物质构成。
与光能、辐射能和电能等能够引发反应不同,催化剂不能引发热力学不允许的反应,而只能使热力学允许的反应加速。光、辐射和电不是物质,故不是催化剂。另外,光照引发的光化学平衡也与热化学平衡不同[6]。
(3) 催化剂的用量少:少数定义将“用量少”作为催化剂的一个特征。实际上催化剂的用量是不固定的,通常而言,催化剂使用量越多,反应速率也越快,呈现线性正比关系[3,12]。而催化剂的用量与催化剂的转换频率(turn-over frequency,TOF)有关,TOF大的,催化剂用量可以相对少一些[13]。但在很多情况下催化剂的用量并不少,即使是实验室中的反应。例如硫酸催化醇脱水制醚和烯烃,硫酸的用量就相当大。而工业上使用催化剂都以千克做单位。因此,“少量”并非催化剂的本质特征,目前多数定义已不再强调少量。
(4) 催化剂加快反应速率。目前,多数专著和教科书都采用“加快”反应速率的表述,但国内依然有不少教科书采用“改变”的说法[5-7]。“改变”意味着可以加快也可以减慢,即认可“负催化剂”的概念。实际上“负催化剂”并不符合催化剂的特征[4],是一个不科学的概念,这一点将在后面做说明。如果排除“负催化剂”,即可将“改变”改为“加快”。这种变化是一个趋势,也与IUPAC的新定义一致。
实际上,升温、加大浓度、改换反应介质[5,13]也可以加快反应,所以加快反应并非催化剂独有的特征。采用“增大反应速率常数”的说法可以排除浓度的影响,但依然不能排除温度和反应介质的影响。只有强调通过改变反应途径加快反应速率,才是催化剂的独有特征(参见(8)和(9)的讨论)。
(5) 催化剂在反应前后质量不变。目前多数催化剂定义都指明催化剂的质量(数量)在反应前后不发生变化。事实上,有很多催化剂会在使用过程中发生损耗,如Pd催化剂和AuPd二元催化剂催化甲酸分解制氢,在使用过程中会发生Pd的溶解损失,回收的催化剂质量会明显减少[14,15]。另外,有的催化剂在反应过程中会被消耗,如碱催化的皂化反应,NaOH会变转化为脂肪酸盐[3]。因此,该性质仅对“理想”的催化剂适用[16]。
(6) 催化剂的化学性质在反应前后不发生变化。通常认为催化剂在使用前后其物理性质(如颗粒尺寸、金属光泽等)会发生改变,但其化学性质不变。但近年的研究表明[2],催化剂在使用过程中会发生重结晶、晶面重构等现象,多元催化剂还会出现偏析、相分离和活泼组分流失等变化;另外,催化剂还会发生中毒,如Pt、Fe催化剂表面可能形成硫化物、砷化物、磷化物等[3]。这些过程会导致催化剂的化学组成和性质的改变,致使催化活性降低,甚至完全失活。虽然从催化剂的作用机理来说,催化剂的化学性质的确不发生改变,但IUPAC的新定义已经不再强调这一点。
(7) 催化剂参加反应。Berzelius曾认为催化剂不参与反应[1],但现今催化剂参与反应已是公认的事实。多数催化理论均认为催化剂能够与反应物发生作用,形成中间体或者过渡态,而后分解形成产物并游离出催化剂[5,6]。
从这个角度说,催化剂既不同于浓度、温度、压强、反应介质等反应条件,又不同于反应物和产物。菲利波夫明确将催化剂列入反应条件范畴[3],之前的定义多数倾向于将催化剂作为反应外部条件看待。这是因为催化剂并非反应必须的,很多催化反应即使没有催化剂也可以或快或慢地进行;另一方面,催化剂是可以更换的,即可以采用不同的催化剂催化同一反应,这显然与反应物有明显区别。
但在 IUPAC的新定义中把催化剂明确列为反应物和产物[4]。关于这一表述的合理性,将在(11)中做进一步的讨论。
(8) 催化剂改变反应途径。这也是目前的共识[5-10]。对多相催化而言,反应过程增加了吸附、活化、脱附等过程,形成了催化剂-反应物、催化剂-过渡态、催化剂-产物等一系列中间状态;对均相催化或者酶催化反应则形成配合物或者加合物等中间体。
催化剂与反应物形成复合物是催化剂的重要特征,而“负催化剂”并不具备该特征。这是“负催化剂”概念不合理的一个重要体现。
(9) 催化剂可以降低反应的活化能。这是催化剂的重要特征之一。而“负催化剂”并不存在增加反应活化能的问题,因为此时反应会按照具有较低活化能的非催化方式进行[8],因此“负催化剂”不具备催化剂改变活化能的特征。
此外,通过改变反应介质的方法改变反应物、中间体或过渡态、产物的溶剂化状态,也可以改变反应的活化能[5,13]。通过改变电极电势,也可以改变电化学反应的活化能[17]。而反应介质和电极电势都不可以视为催化剂。所以,降低反应活化能并非催化剂的独有特征,必须与改变反应途径并列才能构成催化剂的独有特征。
(10) 催化剂不改变反应总的吉布斯自由能变化。该特点有不同的表述,包括催化剂不改变反应的方向、催化剂不改变反应的限度、催化剂不影响化学平衡等。采用吉布斯自由能变化进行表述,可以同时体现反应的方向和限度,而且更加量化和准确[4,8]。事实表明,当催化剂用量比较大时有可能会导致平衡的移动,因此用平衡来表述不尽合理[3]。
需要说明的是,如果将催化剂作为条件,则无论是否引入催化剂,反应的始态和终态都不变,因此保持不变(见式(2))。而如果将催化剂作为反应物和产物,虽然催化剂的可以相互抵消(见式(1)),但抵消必须以忽略催化剂与参加反应的物质之间所产生的化学、物理作用为前提[3]。相对而言,将催化剂作为条件更易理解反应总的吉布斯自由能变化不变。
(11) 催化剂与参与反应的物质之间不存在计量关系。很少有教科书或者专著提及该特点,但菲利波夫特别强调了这一点[3]。
实际上,催化剂的用量并非固定不变的,而是可以任意改变的,故催化剂与反应物、产物之间并不存在如式(1)所示的计量关系,也不需要参与反应式的配平,与通常意义上的反应物和产物之间存在计量关系明显不同。用这一点性质说明催化剂不宜被视为反应物和产物是比较有说服力的。
另外,可以用各物质的浓度变化分别表示反应速率,而这些反应速率之间的比例与该物质的计量系数有关[5,6],存在

而催化剂的浓度不随时间发生变化,因此不能以它的浓度变化来表示反应的速率。这是催化剂不同于反应物和产物的又一重要特征。
关于反应的速率方程,可以讨论如下。
对式(1),其速率方程的一般形式可以写成

当催化剂浓度维持不变时,式(4)可以改写为

式(5)也是式(2)的速率方程。实际上式(2)的速率方程也可能具有式(4)的形式,此时在计量方程中不出现而在速率方程中出现的物质C就是催化剂。
将催化剂写入反应式(式(1)),可以自然而然地将催化剂写入速率方程,便于研究催化剂浓度对反应速率(而非速率常数)的影响,这是将催化剂看作反应物或者产物的方便之处。
由于在体系中加入任何不参与反应的物质也不与反应物和产物具有计量关系,因此该特征并非催化剂独有。
(12) 不少教科书还将具有选择性作为催化剂的本质特征[5-7]。但因为吸附剂也具有选择吸附的性质,所以选择性并非催化剂所特有。
从上述分析可以发现,在全部12个性质中,(4)、(6)、(7)、(8)、(9)、(10)是催化剂比较显著的特征,而(1)、(2)、(3)、(5)、(11)和(12)并非催化剂的本质特征。结合前人的思考和上述分析,我们建议将催化剂定义为:
可以改变反应途径,增加反应速率而又不影响反应总的吉布斯自由能变化的物质。
该定于与IUPAC的新定义最为接近,不同点就在于强调了“改变反应途径”,删除了“既是反应物又是产物”的表述。
2 负催化剂与抑制剂
在明确了催化剂的特征之后我们不难发现,所谓“负催化剂”除了减缓反应发生的速率外,没有任何一点与“催化剂”概念相关。所谓“负催化剂”,从其作用原理看,主要有以下4种情况:
(1) 遮蔽或者直接吸收外界能量,避免反应物被激发活化而发生反应。如在轮胎中加入炭黑、ZnO和TiO2等物质,可以遮蔽或者吸收紫外线,防止紫外线破坏橡胶的C=C键而使橡胶老化。
(2) 消耗反应过程形成的活性中间体,使反应受阻。如在食用油中加入没食子酸正丙酯可以抑制食用油的酸败。这是最常用的“负催化剂”的实例[16]。但没食子酸正丙酯的主要作用是与氧自由基反应从而阻断自由基链传递,其本身会不断被消耗而没有再生,根本不具备催化剂的基本特征。另一种作用方式是淬灭,即加入的物质通过吸收反应物已吸取的能量,使反应物失活而无法反应。
(3) 阻碍反应物之间的接触。这在金属腐蚀防护领域经常使用。如缓蚀剂(inhibitor)的主要作用机制是在金属表面吸附、形成沉淀或者生成转化膜,阻挡腐蚀性介质与金属的接触,从而降低腐蚀速率[6]。
(4) 占据催化剂活性位置,使其不能与反应物结合。例如在 Pt催化剂中加入 Pb等具有一定毒性的物质,可以掩盖活性高的部分,减少副反应并减缓催化剂中毒[3,18]。酶催化反应中的抑制作用就是抑制剂与反应物竞争酶的活性位点的结果[5]。
从本质上说,催化剂的作用主要体现在以下2个方面:
(1) 通过改变反应途径改变活化能,导致指前因子A和活化能Ea改变从而增加速率常数k;
(2) 催化剂的反应级数通常为+1,这主要针对均相催化和酶催化,对多相催化该关系通常不存在。
负催化剂的典型特征是其反应级数为−1,但它并不影响反应的速率常数和活化能。上述3种情况没有一个体现与反应物的直接作用,也没有一个是针对反应指前因子和活化能的。就其作用机制而言,用“抑制剂”描述其作用显然比“负催化剂”更加科学准确。因此,IUPAC在其新定义中摈弃“负催化剂”的概念而使用抑制剂概念是非常合理的。
3 结语
通过对催化剂的概念和特征的比较和分析,明确了催化剂最重要的特征包括改变反应途径、增大反应速率常数、不影响总反应的吉布斯自由能变化,对正确定义催化剂给出了建议。需要强调的是,在教材修订时,宜采用抑制剂概念替代“负催化剂”。
猜你喜欢
特产研究(2024年1期)2024-03-12 05:40:56
天然产物研究与开发(2019年10期)2019-11-05 10:12:44
中学化学(2017年5期)2017-07-07 08:40:47
数学物理学报(2017年2期)2017-06-05 09:12:33
新校园·中旬刊(2017年2期)2017-05-12 02:12:41
课程教育研究·新教师教学(2016年26期)2017-04-10 21:18:57
中学化学(2016年4期)2016-05-30 16:20:37
化工进展(2015年3期)2015-11-11 09:05:36
中学化学(2014年1期)2014-04-23 08:59:04
河北医科大学学报(2011年1期)2011-03-25 10:15:30
