
“核磁共振法定量测定酚氨咖敏药片中各组分”实验的改进
2021-04-09 11:15:24陈文学刘莎莎李会香
大学化学 2021年2期
陈文学,刘莎莎,李会香
复旦大学化学系,国家级化学实验教学示范中心,上海 200433
核磁共振(NMR)技术自问世以来,因其具有样品准备简便、检测快速、重复性好和对样品无破坏性等优势一直深受化学、生物学和医学等领域的专家青睐[1-4]。随着现代科学技术的发展(如计算机科学),以及谱仪场强的不断提高,核磁共振技术的应用和方法研究更是日新月异,它不仅可同步实现定性鉴定和定量分析,而且其检测灵敏度、准确度和精密度等逐渐接近或达到高效液相色谱(HPLC)的定量分析水平。因此,核磁共振检测分析技术和方法已广泛被引入到英国、美国和欧洲等国家药典。
NMR技术有三个重要参数:化学位移、偶合常数和共振峰面积。其中化学位移、偶合常数是结构测定的重要参数,共振峰面积则是定量分析的重要参数。利用 NMR方法进行定量分析的前提条件是:样品中的每个组分中至少有一个或一组特征吸收峰不出现相互重叠;对于1H NMR来说,定量依据为:共振峰面积与产生该共振峰的质子数成正比。
为让学生了解NMR谱仪的工作原理并掌握基于NMR波谱技术对化合物(或药物)进行定性和定量分析的基本方法,本实验中心已为我校化学相关专业的大二本科生开设了“核磁共振法定量测定酚氨咖敏药片中各组分”教学实验[5],其目的主要是服务于具有一定化学背景的化学系、材料系和高分子系等院系的本科学生,其实验课程为“仪器分析和物理化学实验A(上)”。迄今,该教学实验已开设至少满六个学期以上,且每次实验均是单人单次进行和完成的(包括样品制备、NMR上机操作和数据处理等),每次实验时间约3-4课时(半天),学生以分组轮流形式每周均安排本教学实验,每学期完成该实验近200人次。
在该实验中待测药片的样品处理需要水浴(约40 °C)加热以增加组分的溶解度,而NMR谱仪测试时样品温度多为常温(约22 °C),那么在水浴溶解到冷却至室温进行1H NMR检测时是否会有组分析出而影响检测结果?鉴于此,我们拟对本实验进行部分改进和探索:一是移去水浴加热步骤,改用室温直接溶解处理样品。同时为保证药片中各组分能充分溶解,选择合适的药片用量;二是随着谱仪分辨率的提高,增加微量组分马来酸氯苯那敏的含量测定。
1 实验仪器和试剂
1.1 仪器
Bruker AVANCE NEO 400 MHz核磁共振谱仪(Bruker公司,德国),精密电子天平(感量0.1 mg,Sartorius公司,德国),离心机(微型,杭州米欧仪器公司,中国),控温仪(上海羌强仪器设备公司,中国),移液枪(0.2 mL、1.0 mL,北京大龙兴创实验仪器公司,中国),研钵,容量瓶(10.0 mL),烧杯(10.0 mL)。
1.2 试剂和药品
重水和 3-三甲基硅烷-1-丙磺酸钠(CIL公司,英国),氨基比林、对乙酰氨基酚、咖啡因和酚氨咖敏药片均为上海上药信谊药厂出品。
2 实验方法
2.1 实验内容
酚氨咖敏药片,英文名:Paracetamol, Aminophenazone, Caffeine and Chlorpheniramine maleate tablet (PACC)。多为由对乙酰氨基酚组成的复方制剂,性状为白色片。是主要用于医治感冒、发热、头痛、神经痛及风湿痛等症的常见药品。通常酚氨咖敏片包含的主要药效成分为:氨基比林、对乙酰氨基酚、咖啡因和马来酸氯苯那敏。
(1) 氨基比林(aminophenazone)。化学名为1-苯基-2,3-二甲基-4-二甲氨基吡唑酮,为白色或几乎白色结晶性粉末。氨基比林主要起退热、镇痛和抗炎作用。
(2) 对乙酰氨基酚(paracetamol)。化学名为N-(4-羟基苯基)乙酰胺,为白色结晶或结晶性粉末。对乙酰氨基酚也主要起解热和镇痛作用。
(3) 咖啡因(caffeine)。别称:1,3,7-三甲基黄嘌呤,为白色粉末或有光泽的针状结晶。咖啡因属中枢神经兴奋药,能起缓解头痛作用。
(4) 马来酸氯苯那敏(chlorpheniramine maleate)。化学名为 N,N-二甲基-γ-(4-氯苯基)-2-吡啶丙胺顺丁烯二酸盐,为白色结晶性粉末。马来酸氯苯那敏可减轻因过敏引起的呼吸道症状,对中枢神经系统也有轻度抑制作用。
根据实验教材[5]和文献报道[6,7],酚氨咖敏药片中各组分的分子式、分子量和核磁共振氢谱中所对应的主要特征信号峰(s)等信息总结在表1。

表1 酚氨咖敏药片中各组分的核磁共振氢谱的主要特征信号峰
如用内标法进行1H NMR波谱测定。可以选取3-三甲基硅烷-1-丙磺酸钠(DSS钠盐,分子式:C6H15NaO3SSi;分子量为218.32)作为内标物质,并称取一定量酚氨咖敏药品(WS)和内标物(WR),再根据NMR法测定酚氨咖敏片中各主要组分的含量。
定量计算依据就是将样品中某组分指定基团上的质子引起的共振峰(即吸收峰)面积与由内标物质指定基团上的质子引起的共振峰面积进行比较,便可算得样品中该组分的含量,其计算公式如下:

其中,WX和MX分别为样品中氨基比林、对乙酰氨基酚、咖啡因和马来酸氯苯那敏等某组分的质量(含量)和分子量;而WR和MR分别为内标物的质量和分子量;SX和SR分别为样品和内标物的特征峰面积,HX和HR则分别为样品和内标物的特征峰基团中相应的质子个数。
2.2 实验步骤
2.2.1 混合标准样品的配制和回收率测定
准确称取氨基比林112 mg,对乙酰氨基酚104 mg,咖啡因34 mg,内标物(3-三甲基硅烷-1-丙磺酸钠) 68 mg,用重水溶解并定量转移于10.0 mL容量瓶内,用重水定容后备用。
取上述混合标准样品溶液0.5 mL于NMR样品管中,按照核磁共振谱仪使用步骤进行1H NMR谱测定。
2.2.2 样品预处理和药片各组分含量测定
取一片酚氨咖敏药片(约500 mg),在研钵中将药片研成细粉状。
准确称取研细的药片约20.0 mg (WS),置于1.0 mL离心管,加入重水0.6 mL,再加入3-三甲基硅烷-1-丙磺酸钠重水溶液(20.0 mg·mL−1) 0.1 mL。振荡摇匀,然后在室温直接离心分离,并取上层清液0.5 mL于NMR样品管内,同上测定其1H NMR谱。
分别准确称取研细的药片约20.0 mg和40.0 mg (WS),置于1.0 mL离心管,加入重水0.6 mL,再加入3-三甲基硅烷-1-丙磺酸钠重水溶液(20.0 mg·mL−1) 0.1 mL。振荡摇匀后,于40 °C水浴加热10 min左右,再离心分离(转速4000 r·min−1,3 min);最后取其上层清液0.5 mL于NMR样品管内,同上测定其1H NMR谱。
2.3 实验改进
2.3.1 减小待测样品用量,室温测定药片各组分含量
根据药片中各组分在水中的溶解度情况(见表2),在前期实验[5]中,因药片组分含量测定称取的样品量均为40.0 mg左右,那么采用水浴(40 °C)条件主要是为了增加和加速各组分的溶解,但在离心后冷却至室温进行1H NMR测定时,我们推测药片中溶解度相对较小的对乙酰氨基酚组分可能有微量析出。因此,为提高检测的准确度和精密度,本研究将适当减小待测样品用量,并拟改在温和(室温约22 °C)条件下直接测定其药片中各组分的含量,从而与40 °C水浴测定结果进行比较。
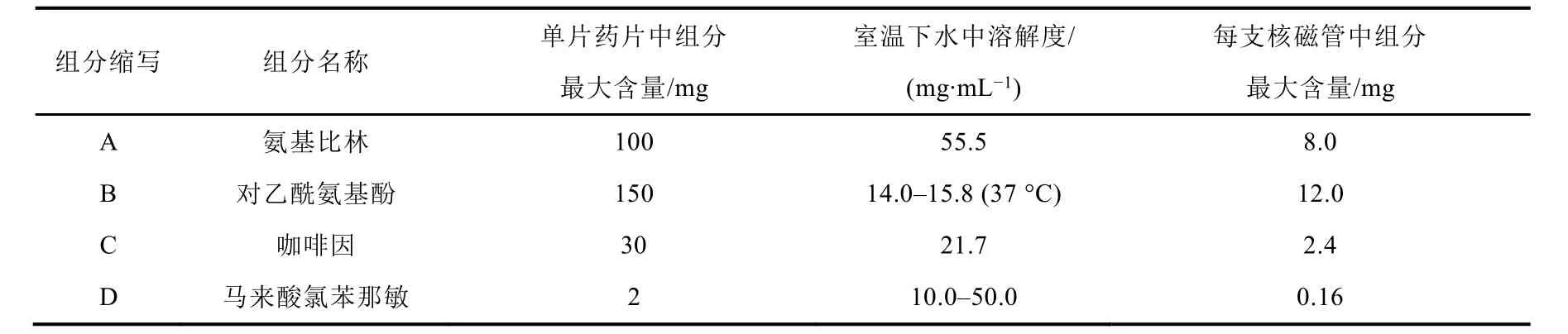
表2 酚氨咖敏片中各组分的含量和溶解情况
2.3.2 增加药片中马来酸氯苯那敏组分含量测定
酚氨咖敏药片属复方制剂,其药品说明书上标明有四种药效组分,先前实验[5]因受核磁共振仪器分辨率(60 MHz)所限只测定了氨基比林、对乙酰氨基酚和咖啡因三种主要组分的含量,本文拟增加含量较小的马来酸氯苯那敏(每片中约含2.0 mg,质量分数约0.4%)组分的含量测定。
图1是约40.0 mg酚氨咖敏药片重水溶液的400 MHz核磁共振氢谱。如图1所示,数字1是内标物DSS的信号峰;数字2-8等信号峰(高场δ2.0-4.0)代表氨基比林、对乙酰氨基酚和咖啡因三组分中甲基的特征信号峰(单峰);数字9是溶剂重水D2O的信号峰;数字10号共振峰(即左上放大8倍的NMR谱图)则为微量组分马来酸氯苯那敏中马来酸的共轭双键上的氢信号(单峰)。

图1 酚氨咖敏药片中各主要组分的400 MHz核磁共振氢谱
3 实验结果与讨论
3.1 待测样品减量和样品处理条件对药片各组分含量测定影响
根据实验教材[5,8]和文献报道[6,7],首先依次称取了约20 mg待测样品18份,并分别在22 °C室温和40 °C水浴(约10 min)条件下用1H NMR方法测定了酚氨咖敏药片中各组分的含量。再依次称取约40 mg待测样品6份,只在40 °C水浴条件下测定其药片中各组分的含量。经多次重复实验,所得的各组分含量见表3。
由表3可知,在待测样品称量均为20 mg时,水浴组(class 2)药片中测定的各组分含量与室温组(class 1)直接测定结果基本一致。同样,如同为40 °C水浴条件时,与低称量组(class 2)相比,高称量组(class 3)药片中测定的各组分含量也几乎同比例增加。

表3 室温22 °C和40 °C水浴时测定酚氨咖敏药片中各组分含量(mg)
此外,上述三组样品在室温放置四天后再行1H NMR波谱测定,结果发现每组样品中各相同组分的含量几乎没有变化(数据未列出),这说明待测样品不仅非常稳定,而且该检测方法重现性很好。
3.2 样品中各组分含量统计分析
为观察水浴条件或药片用量的减小对组分含量测定的影响,本文对上述实验结果依次使用统计软件(SPSS 17.0,美国SPSS公司)中独立T检验方法进行了组间统计分析,结果发现如图2所示:不仅在Class 1和Class 2之间各组分的含量几乎无显著性差别(p> 0.05),而且在Class 2和Class 3之间各组分的含量也几乎无显著性差别(p> 0.05)。这就是说,在当前温和(22 °C室温)条件下,该方法对药片中各组分含量的测定与40 °C水浴条件测定结果是一致的;即使减少待测样品用量,也同样可实现对微量组分马来酸氯苯那敏的准确测定。

图2 室温22 °C和40 °C水浴时测定三组样品中各组分含量(mg)的均值、标准偏差和p值
4 结论
通过实验改进和探索,我们发现待测样品直接在室温下溶解,或减少药片的用量(保障药片各组分在室温下完全溶解),均不会影响1H NMR方法定量测定酚氨咖敏药片中氨基比林、对乙酰氨基酚和咖啡因等三组分含量。除此之外,随着核磁共振谱仪场强的提高,检测限降低,1H NMR方法还可定量测定酚氨咖敏药片中微量的马来酸氯苯那敏组分含量。
总之,1H NMR测定方法具有样品处理简单、用量少,对样品无破坏性,检测快速、重现性好,且无须分离等诸多优势。显然,该方法不仅可同步实现酚氨咖敏药片各组分含量测定,而且其测定速度、准确度和精密度等均接近或达到高效液相色谱的定量分析水平[9,10]。
也许,不久该方法测定酚氨咖敏药片各组分含量还有望被录入《中国药典》。
猜你喜欢
山西化工(2021年5期)2021-01-25 15:00:58
奥秘(创新大赛)(2020年3期)2020-11-28 23:40:32
保健与生活(2020年4期)2020-03-02 02:27:36
作文大王·低年级(2019年4期)2019-05-13 01:44:10
天然产物研究与开发(2018年5期)2018-06-13 03:23:32
发明与创新(2018年6期)2018-04-01 11:11:05
发明与创新·中学生(2018年2期)2018-02-07 18:21:36
中国药业(2014年24期)2014-05-26 09:00:21
中国药业(2014年19期)2014-05-17 03:12:22
济源职业技术学院学报(2014年3期)2014-02-28 02:35:42
