
HCN与HNC异构化反应活化能的量化计算实验
2021-04-09 11:15许秀芳
大学化学 2021年2期
许秀芳
南开大学化学学院,天津 300071
活化能是指化学反应中将反应物分子活化成活化分子(亦称为活化络合物或过渡态)所需的能量[1],或者是全部活化分子(活化络合物或过渡态)的平均能量与全部反应物分子平均能量之差[2]。化学反应活化能是化学动力学中的重要参数,与化学反应速率的大小密切相关,活化能越低,反应速率越快,因此活化能在化学及化工诸多领域都有着广泛的应用。常用的测定化学反应活化能的方法主要是通过测定不同温度下化学反应的速率常数,进而利用Arrhenius公式求算反应的Arrhenius活化能。当活化分子(活化络合物或过渡态)转化为产物的速率常数不随温度而变或指前因子与温度无关时,Arrhenius活化能即为反应活化能[1]。而测量化学反应速率常数的实验方法主要有化学方法和物理方法。化学方法是采用骤冷、稀释或加入阻化剂等方法使反应停止,进而通过化学分析法测定反应中各物质的浓度变化来求算速率常数。物理方法则是利用物质的物理性质的变化测定反应中各物质浓度的变化从而测算速率常数,其优点是在测定时不会干扰或破坏化学反应的进行。通常用于求算速率常数的物理方法包括电导法、旋光度法、分光光度法、碘钟法、差热分析法、熔融指数法、基团估算法等[3-6]。
在本文中,我们介绍一个利用量化计算获得化学反应活化能的计算化学实验。该实验运用量子化学软件Gaussian 09[7]及其配套的可视化软件GaussView和文本编辑器UltraEdit,通过量子化学方法计算HCN⇌HNC异构化反应中反应物、过渡态和产物的能量,从而求算反应的活化能。HCN⇌HNC异构化反应作为一个非常典型的单分子反应受到了广泛关注,实验上通常用激光诱导的方法研究该反应的动力学过程[8,9],理论上对该反应的研究涉及到用各种理论方法研究异构化概率、反应物、产物的振动能级、反应的过渡态和活化能等等[10-14]。该异构化反应看似简单,但它包含了与化学反应有关的很多基本问题,如稳定反应物和产物的确定,过渡态结构的构造,正逆反应活化能,反应速率常数,反应热等等。该实验使用Gaussian 09软件进行量化计算,实验过程涉及初始构型构建、输入文件编制、几何构型优化、过渡态搜索、频率计算、能量计算、IRC (Intrinsic Reaction Coordinate,即內禀反应坐标)[15]计算等计算化学主要步骤,具有较高的实用性。另外,选取的分子结构简单,计算量较小,耗时较短,能够保证在规定课时内完成实验内容。
1 实验目的
(1) 用量子化学计算方法求算在101325 Pa和298.15 K下HCN⇌HNC反应中反应物、过渡态和产物的能量,从而求算反应的活化能。
(2) 掌握 Gaussian 09计算化学软件及其配套的可视化软件 GaussView和文本编辑器 UltraEdit的基本使用方法。
(3) 学习使用密度泛函(DFT)方法优化分子结构、搜索过渡态、做IRC计算和计算物质的能量的基本过程,了解输出文件中与能量相关的各参数的意义。
2 实验原理
Gaussian软件是目前使用极为广泛的一款量子化学计算软件。该软件由诺贝尔奖获得者波普尔等人于1970年开发,经过四十多年的不断发展和完善,目前最新的版本是Gaussian 16,使用较为广泛的版本是 Gaussian 09。Gaussian软件通常包括用于计算的 Gaussian和用于图形界面处理的GaussView两个组件。其中Gaussian软件涵盖了从头算方法、密度泛函方法、半经验方法、分子力学方法等多种计算方法,可以计算物质的结构、频率、能量、光谱特征等多种参数,具有操作简单、功能强大的特点。在本实验中,需要计算的是101325 Pa和298.15 K下,反应物(HCN)、过渡态(TS)和产物(HNC)的电子能、焓和吉布斯自由能,进而计算HCN⇌HNC反应的活化能、活化焓和活化吉布斯自由能。
3 实验设备
Gaussian 09软件,GaussView 5.0软件,UltraEdit软件,CYLview v1.0软件,联想台式电脑 (i5-4460 4G 500G)。
4 实验步骤
本实验采用Gaussian 09中的B3LYP/6-311+G(d,p)密度泛函方法计算HCN⇌HNC反应中反应物、过渡态和产物的能量,从而求算反应的活化能。具体步骤包括:
(1) 构建初始构型。通过GaussView软件构建反应物(HCN)和产物(HNC)的初始构型,这两个分子的初始构型均为直线型(如图1(a)和图1(b)所示)。基于这两个分子的构型,我们猜测这两个分子异构化反应的过渡态应该具有C-H键正在断裂和N-H键正在形成的结构,因此C、H、N原子形成三角形结构,且其中的C―H键和N―H键以及C―N键均比正常键长值要长。基于以上考虑,我们搭建了初猜过渡态TS的结构如图1(c)所示。

图1 用GaussView软件构建的反应物(HCN)、产物(HNC)和过渡态(TS)的初始几何构型
(2) 编制输入文件。根据现有的计算条件、模型的大小以及所要解决的问题,选择可行的计算方法,采用文本编辑器UltraEdit编制输入文件。在本实验中,采用Gaussian 09中的B3LYP/6-311+G(d,p)密度泛函方法计算各物种的能量。B3LYP是采用广义近似梯度的杂化密度泛函之一。其中,6-311+G(d,p)是基组的名称,表示内层轨道用 6个高斯函数(GTO)拟合一个 Slater函数(STO),然后用这个STO拟合一个原子轨道;价层轨道则分为内、中、外轨,内轨用3个GTO拟合一个STO,中轨和外轨分别用1个GTO拟合一个STO,然后用这三个STO拟合一个价层原子轨道,并且在6-311G基组的基础上给轻原子添加了p轨道极化函数,同时给重原子添加了d轨道极化函数和弥散s和p函数。并且该实验需要用B3LYP/6-311+G(d,p)方法对所计算的分子进行构型优化和频率分析,从而得到准确的能量数据。因此需要在计算执行路径行使用关键词opt和freq,其中opt即是对相应的分子进行构型优化,freq即是对相应的分子进行频率分析。其中,用UltraEdit编制的对HCN分子进行计算时的输入文件(通常以gjf为后缀名)和说明如表1所示。

表1 B3LYP/6-311+G(d,p)方法计算HCN分子的输入文件及各部分的名称和含义
同理,用B3LYP/6-311+G(d,p)方法对HNC分子进行计算时的输入文件如表2所示。

表2 B3LYP/6-311+G(d,p)方法计算HNC的输入文件及各部分的名称和含义
(3) 过渡态搜索,即优化过渡态结构。
过渡态结构优化的算法有三种:TS,QST2和 QST3。其中 TS算法用的关键词为 opt =(ts,calcfc,noeigen),该算法需要输入初始猜测的过渡态结构。QST2算法用的关键词为 opt =(qst2,calcfc,noeigen),该算法需要输入反应物附近和产物附近的两个结构。QST3算法用的关键词为opt = (qst3,calcfc,noeigen),该算法需要输入反应物、产物和初猜的过渡态三个结构。在此,我们选择TS方法优化过渡态,所需的初猜过渡态结构已经在第一步用GaussView软件搭建好。用UltraEdit编制的过渡态搜索输入文件如表3所示。
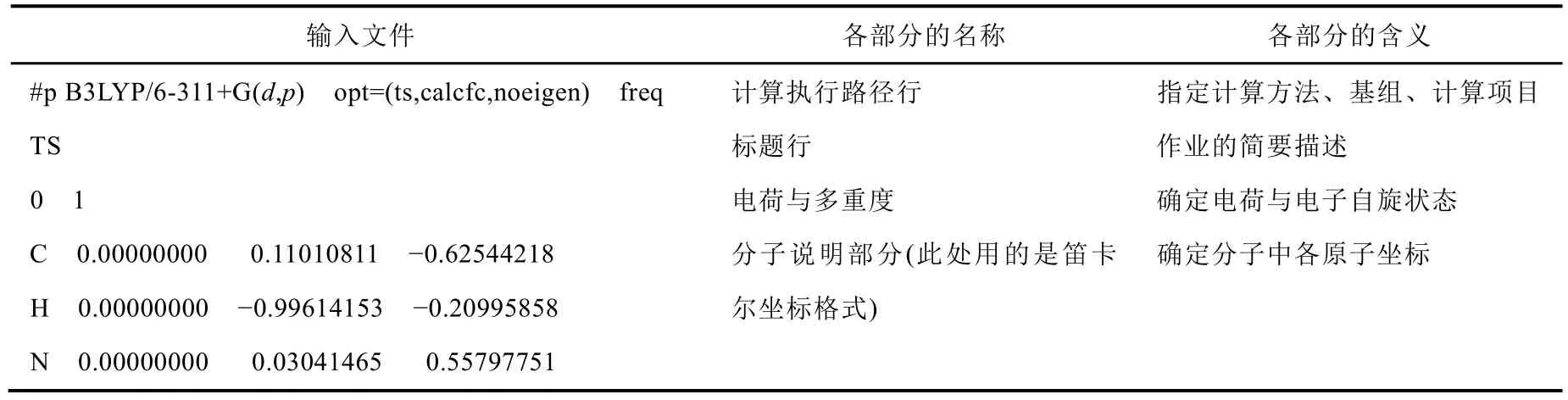
表3 过渡态搜索的输入文件及各部分的名称和含义
(4) 量化计算。启动Gaussian 09,使用“File”下拉菜单“Open”命令将编制好的输入文件带入Gaussian 09进行构型优化、振动频率计算和能量计算等,获得HCN、HNC分子及过渡态TS的平衡几何构型、振动频率和各种热力学能量。计算结果显示HCN和HNC的全部振动频率为实频而无虚频,过渡态TS只有一个虚频−1127.27 cm−1。通过用GaussView显示过渡态的虚频率所对应的振动模式以及进一步做IRC计算,我们确认该过渡态结构是连接HCN和HNC的正确过渡态。IRC计算的输入文件如表4所示,表中的笛卡尔坐标是经过结构优化得到的过渡态的坐标。IRC计算将得到从过渡态出发连接势能面上相邻两个极小点的最低能量途径,我们将通过查看这条最低能量途径上的这两个极小点的结构来确认过渡态是否为正确的过渡态。

表4 IRC计算的输入文件及各部分的名称和含义
IRC计算出的內禀反应坐标如图2所示,此图表明搜索到的过渡态确实连接反应物 HCN和产物HNC,由此可以确认该过渡态是正确的过渡态。
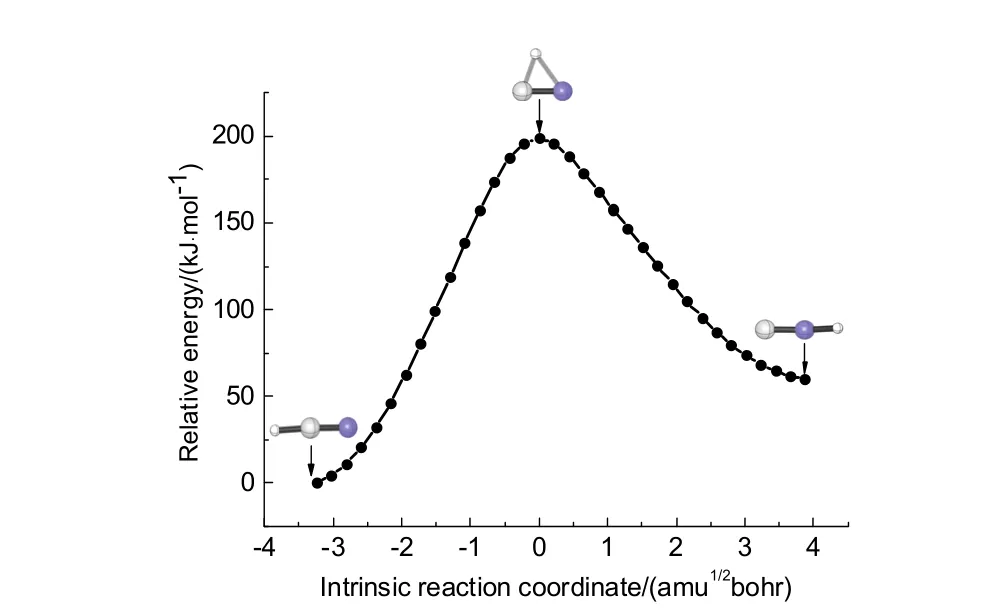
图2 B3LYP/6-311+G(d,p)方法计算所得到的内禀反应坐标(IRC)
为了帮助学生进一步掌握以上操作步骤,图3给出了以上操作步骤的图示说明。
图3 操作步骤图示
(5) 计算结果的分析和整理。计算结束后用GaussView或UltraEdit软件打开*.out输出文件查看全部的计算结果。对计算结果进行分析和整理,一般包括构型描述、能量分析、轨道组成分析、电荷和成键分析等,提取有用的信息。
5 数据处理
(1) 在CYLview v1.0中打开*. out格式的输出文件,可以得到B3LYP/6-31G(d,p)方法优化出的反应物(HCN)、过渡态(TS)和产物(HNC)的三维结构如图4所示。
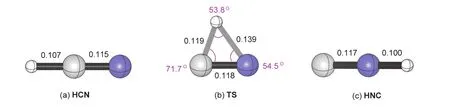
图4 优化得到的反应物(HCN)、过渡态(TS)和产物(HNC)的稳定几何结构
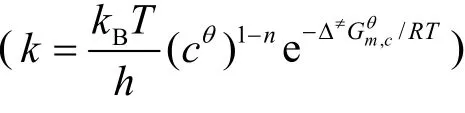
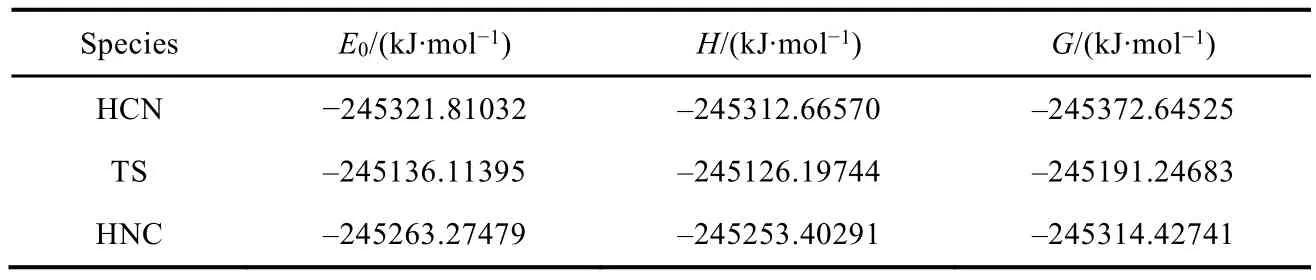
表5 B3LYP/6-311+G(d,p)方法计算出的反应物(HCN)、过渡态(TS)和产物(HNC)的电子能E0、焓H和吉布斯自由能G a, b
根据表5中的输出结果可以计算正向反应活化吉布斯自由能和逆向反应活化吉布斯自由能分别为:正向反应活化吉布斯自由能 ΔG≠(forward) =GTS−GHCN= −245191.24683 − (−245372.64525) =181.4 kJ·mol−1; 逆向反应活化吉布斯自由能 ΔG≠(reverse) =GTS−GHNC= −245191.24683 −(−245314.42741) = 123.2 kJ·mol−1。
另外在输出文件里,“Thermochemistry”部分中的“Sum of electronic and zero-point Energies”项即是对应分子的电子能,此电子能包含了零点振动能;“Sum of electronic and thermal Enthalpies”项即是对应分子的焓。依照以上活化吉布斯自由能的计算方法,我们也可以计算出活化能和活化焓。
为了直观地显示某反应的机理及所需的活化能,通常给出该反应的势能剖面图。HCN和 HNC的异构化过程的吉布斯自由能的势能剖面图如图 5所示,该图采用 ChemBioOffice软件包中的ChemBioDraw绘制。从图5中可以直观地看出,HCN比其异构体HNC稳定58.2 kJ·mol−1。HCN异构化为HNC所需活化吉布斯自由能为181.4 kJ·mol−1,比HNC异构化为HCN所需活化吉布斯自由能(123.2 kJ·mol−1)高 58.2 kJ·mol−1,即 HNC 更容易异构化为 HCN。该异构化反应的正、逆活化吉布斯自由能都比较高,说明此异构化反应在通常条件下不容易发生。
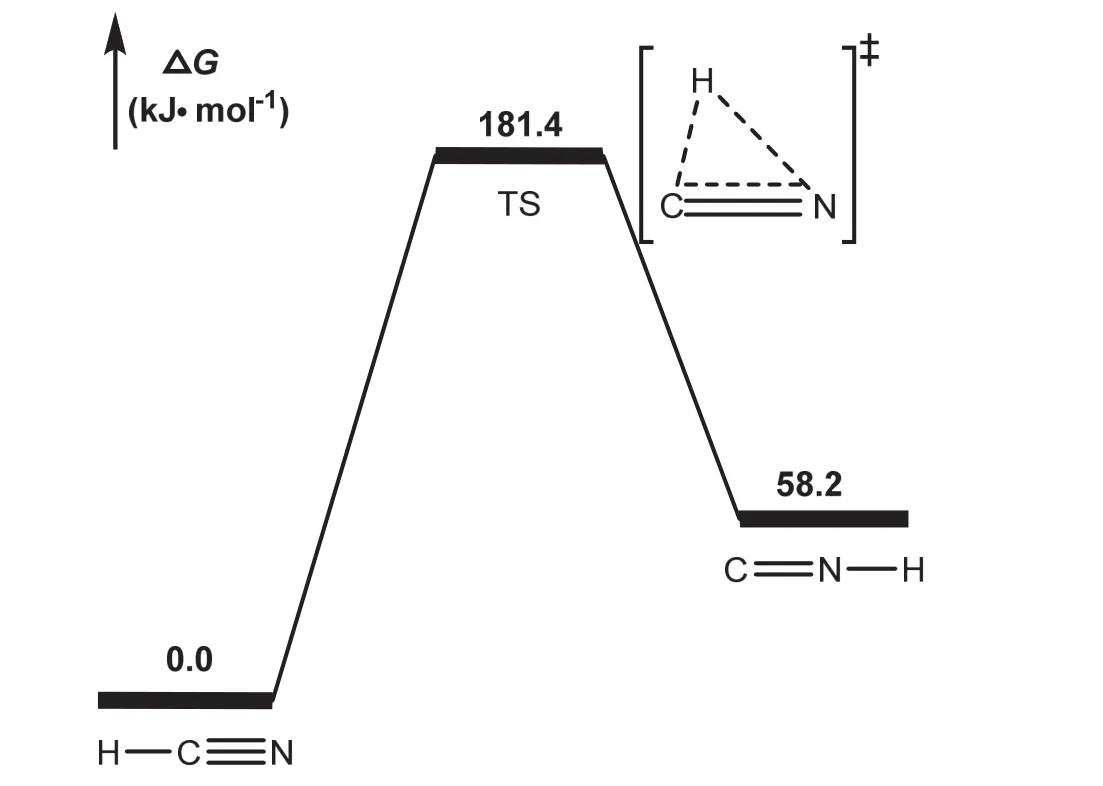
图5 HCN和HNC的异构化过程的势能剖面图
需要说明的是,本实验的计算对象为真空中分子,因此计算的结果也有可能与实验测定值之间存在微小的差异。另一方面,量子化学计算的精度取决于所用的方法和基组,不同方法和基组得到的计算结果在某些情况下差异会很大,应结合实际体系和所具备的实验条件选择适合的方法和基组。
6 思考题
(1) B3LYP/6-31G(d,p)方法计算出的HCN⇌HNC异构化反应的正、逆反应活化吉布斯自由能分别是多少 kJ·mol−1?以上计算出的反应活化吉布斯自由能数值表明该异构化反应在常温常压下容易发生吗?
(2) 用艾琳公式和以上计算得到的活化吉布斯自由能计算该异构化反应的正、逆反应的速率常数。
(3) 使用催化剂可以改变反应机理,从而降低反应的活化能。请通过思考或查阅文献提供一种能够降低此反应活化能的方法并给出相应的反应机理。
(4) 用Gaussian 09 软件和B3LYP/6-31G(d,p)方法计算CH3CN⇌CH3NC异构化反应的过渡态和正、逆反应活化焓和活化吉布斯自由能。
7 实验时间安排
本实验是面向大三下学期或大四上学期,已学习过物理化学课程中量子化学和统计力学相关知识的学生开设的探索性实验。实验用时 4课时,具体为:简要介绍量子化学计算的相关知识,1课时;介绍Gaussian 09、GaussView 5.0、UltraEdit软件的使用方法,如何编制输入文件以及如何查看输出文件,2课时;上机计算并完成数据的计算与整理,1课时。课后学生独立完成实验报告和思考题。
8 实践效果
笔者在计算化学教学中,一直坚持理论教学和实践教学相结合的教学方法,并将“讲一练二考三”的模式引入到计算化学教学中,增加实践练习在教学中的比例,还尝试了利用举办计算化学知识技能竞赛的方式促进计算化学教学[16]。开设“HCN与HNC异构化反应活化能的量化计算”这一计算化学实验的目的也是希望学生能通过量化计算的方法在一定程度上解决有机化学、无机化学、物理化学、应用化学、化学工程等诸多领域都会涉及的化学反应活化能求算的问题。很多学生表示,通过本实验的操作,掌握了一种较为简便有效地求算化学反应活化能的方法,对于将来的科研工作中通过计算和比较不同反应路径的活化能从而确定反应机理很有帮助。另外,通过这一实验和其它类似的计算化学实验的开设,不但加深了学生对计算化学知识的理解,而且提高了学生对学习计算化学的兴趣,有些学生在后来的本科毕业论文(设计)和研究生阶段选择了与计算化学相关的课题。同时一些选择有机化学、无机化学、材料化学方向课题的学生也基于计算化学实验的经历,有意识地将计算化学方法应用于自身的科研工作中。值得一提的是,我们也将这一计算化学实验在工科院校化学或化工相关专业的学生中进行了教学实验的尝试。实践表明,即便没有计算化学基础的学生也均能比较顺利地完成实验,取得令人满意的结果。
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
石油石化绿色低碳(2019年6期)2019-01-14
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
大学化学(2016年4期)2016-07-27
化工进展(2015年3期)2015-11-11
华东理工大学学报(自然科学版)(2015年3期)2015-11-07
学园(2015年5期)2015-10-21
中国粮油学报(2014年8期)2014-02-06
