
芦荟的HPLC指纹图谱建立、化学模式识别分析及其含量测定
2021-04-07 03:26严雅慧李淑萍热依木古丽阿布都拉阿吉艾克拜尔艾萨
天然产物研究与开发 2021年3期
严雅慧,李淑萍,热依木古丽·阿布都拉,阿吉艾克拜尔·艾萨*
1中国科学院大学,北京 100049;2中国科学院干旱区植物资源化学重点实验室中国科学院新疆理化技术研究所,乌鲁木齐 830011
芦荟为重要的可食用的药材,是百合科芦荟属多年生常绿肉质草本植物,性寒味苦,古方中记载其具有清肝热、通便、散结、疗癣等作用。芦荟总共有500多个品种,其中库拉索芦荟(Aloevera(L.) Burm.f.)是最常用的药用品种,其制品也广泛应用于食品(包含保健食品)、药品、化妆品等多个领域[1]。近年来,国内外芦荟的研究工作主要集中在品种的对比分析[2,3]、分离纯化[4,5]、质谱分析[6,7]、含量测定[8,9]和药理作用[10,11]等方面。研究证明,芦荟的主要活性成分为蒽酮类、蒽醌类和色酮类化合物,此外还有多糖类及糖苷、氨基酸、有机酸等化学成分[12,13],具有抑菌、消炎、抗肿瘤、抗病毒、抗过敏等活性[14-16]。
目前,指纹图谱结合化学模式识别是评价中药材质量的有效手段。指纹图谱主要关注药材的整体性,建立共有模式,识别共有峰,能够客观、全面揭示药材中可能的活性成分种类和数量;而化学模式识别技术则强调的是药材之间差异性,通过对指纹图谱数据信息的整合和校正,进一步缩小药材活性成分的范围;两者结合可实现药材的整体描述到个体分析的过程,可更加准确、客观地反映中药的质量差异[17,18]。芦荟药材主要药用成分以芦荟苷、芦荟大黄素等化合物为主[19],但功效成分及其含量随药材产地、生长环境、土壤及采摘季节等因素而不同,给芦荟药材品质及安全性的评价带来了一定的困难。此外,《中国药典》(2020版)[20]中芦荟药材的含量测定项目以芦荟苷含量来制定,指标成分单一,难以控制药材质量。因此,在生产和开发芦荟药材相关制剂之前,对芦荟药材进行指纹图谱和结合化学模式识别研究是必不可少的。
本研究采用高效液相色谱(HPLC)法建立12批次不同产地的芦荟药材的指纹图谱,并结合相似度分析、聚类分析、主成分分析(PCA)和正交偏最小二乘判别分析(OPLS-DA)研究其化学组分特征,筛选出能影响药材质量差异的标志物,并用相同的方法测定12批芦荟药材中6个标志物(芦荟新苷D、芦荟苷A、芦荟苷B、7-O-甲基芦荟新甙A、芦荟苦素和芦荟大黄素)的含量,旨在从生产源头确保药材品质,为芦荟药材质量控制和标准完善提供科学依据。
1 实验仪器与材料
1.1 仪器
美国Water 2695液相色谱仪(包括自动进样器,DAD紫外检测器;Empower色谱工作站,四元梯度泵,在线过滤器,柱温箱);BT25S型电子分析天平(赛多利斯科学仪器(北京)有限公司);SQP型电子分析天平(赛多利斯科学仪器(北京)有限公司);SK7210 HP型超声波(上海科导超声仪器有限公司)。
1.2 试剂与材料
对照品:芦荟苦素(批号:19011601)、芦荟新苷D(批号:19091701)、芦荟苷B(批号:19030501)、芦荟苷A(批号:19030204)、芦荟大黄素(批号:19010803)、7-O-甲基芦荟新甙A(批号:19101301)均采购于上海纯优生物科技有限公司,以上对照品纯度均 ≥ 97.5%。
甲醇和乙腈均为色谱纯(Merck公司,Darmstadt,德国);磷酸和甲醇为分析纯(天津市光复精细化工研究所);娃哈哈纯净水(杭州娃哈哈集团有限公司);其他所有试剂均为分析纯。
12批芦荟药材来源于广东、广西、海南和四川4个省份,其中S1、S2、S7来源于四川省,S10~S12来源于广西省,S3~S5来源于广东省,S6、S8、S9来源于海南省。经中国科学院新疆理化技术研究所刘戈宇研究员鉴定为库拉索芦荟Aloevera(L.) Burm.f.的浓缩物。
2 实验方法与结果
2.1 HPLC指纹图谱建立
2.1.1 样品制备
参照中国药典方法[25]略微改动,取芦荟药材粉末(过5号筛)50 mg,精密称定,精密加入50%甲醇75 mL,称定重量,超声处理(40 kH,700 W,25 ℃)20 min,放冷,再称定重量,用50%甲醇水补足减失的重量,摇匀,分析前过0.45 μm微孔滤膜,备用。
2.1.2 对照品溶液制备
精密称取各对照品适量,精密称定,置于棕色容量瓶中,用甲醇溶解并定容,配制成对照溶液:芦荟苦素(316 μg/mL)、芦荟新苷D(524 μg/mL)、7-O-甲基芦荟新甙A(228 μg/mL)、芦荟苷B(508 μg/mL)、芦荟苷A(506 μg/mL)、芦荟大黄素(436 μg/mL),精密量取各对照溶液按一定比例混合配制成不同浓度的6份混合对照溶液,摇匀,过0.45 μm微孔滤膜备用,-20 ℃避光保存。
2.1.3 色谱条件
HPLC色谱柱:Gemini C18色谱柱(250 mm×4.6 mm,5 μm,Phenomenex Inc.,CA,USA),流动相溶剂A:0.3%(V/V)磷酸水溶液,B:乙腈,C:甲醇,流速是1 mL/min,进样10 μL,柱温35 ℃,检测波长为254 nm,最佳梯度洗脱如下:0~8 min,88%A+10%B+2%C;8~10 min,74%A+10%B+16%C;10~45 min,58%A+10%B+32%C;45~80 min,55% A+10%B+35%C。
2.2 指纹图谱的方法学考察
2.2.1 精密度
取编号S1的样品,按“2.1.1”项下处理,按“2.1.3”项下色谱条件连续进样6次,记录液相色谱图。以15号色谱峰为参照峰,计算各共有峰相对保留时间的RSD<0.19%,相对峰面积的RSD<0.89%(n=6),表明仪器精密度良好。
2.2.2 稳定性
取编号S1的样品,按“2.1.1”项下处理,按“2.1.3”项下色谱条件分别在0、2、4、8、12、18、24、48 h进样,记录液相色谱图。以15号色谱峰为参照峰,计算各共有峰相对保留时间的RSD<0.87%,相对峰面积的RSD<2.67%(n=8),表明样品溶液在48 h内稳定性良好。
2.2.3 重复性
取编号S1的样品,精密称取6份,按“2.1.1”项下处理,按“2.1.3”项下色谱条件进行测定,记录液相色谱图。以15号色谱峰为参照峰,计算各共有峰相对保留时间的RSD<0.42%,相对峰面积的RSD<2.66%(n=6),表明方法重现性良好。
2.3 指纹图谱的生成
12批芦荟药材按“2.1.1”方法制备,再按“2.1.3”色谱条件进样测定,记录液相色谱图。采用《中药色谱指纹图谱相似度评价系统(2012 A版)》,以S1为参照图谱,采用中位数法、时间窗宽度设为0.1 min建立对照图谱,用多点校正Mark峰匹配,建立12批芦荟药材样品的指纹图谱。叠加指纹图谱、参照图以及共有峰标记均见图1。
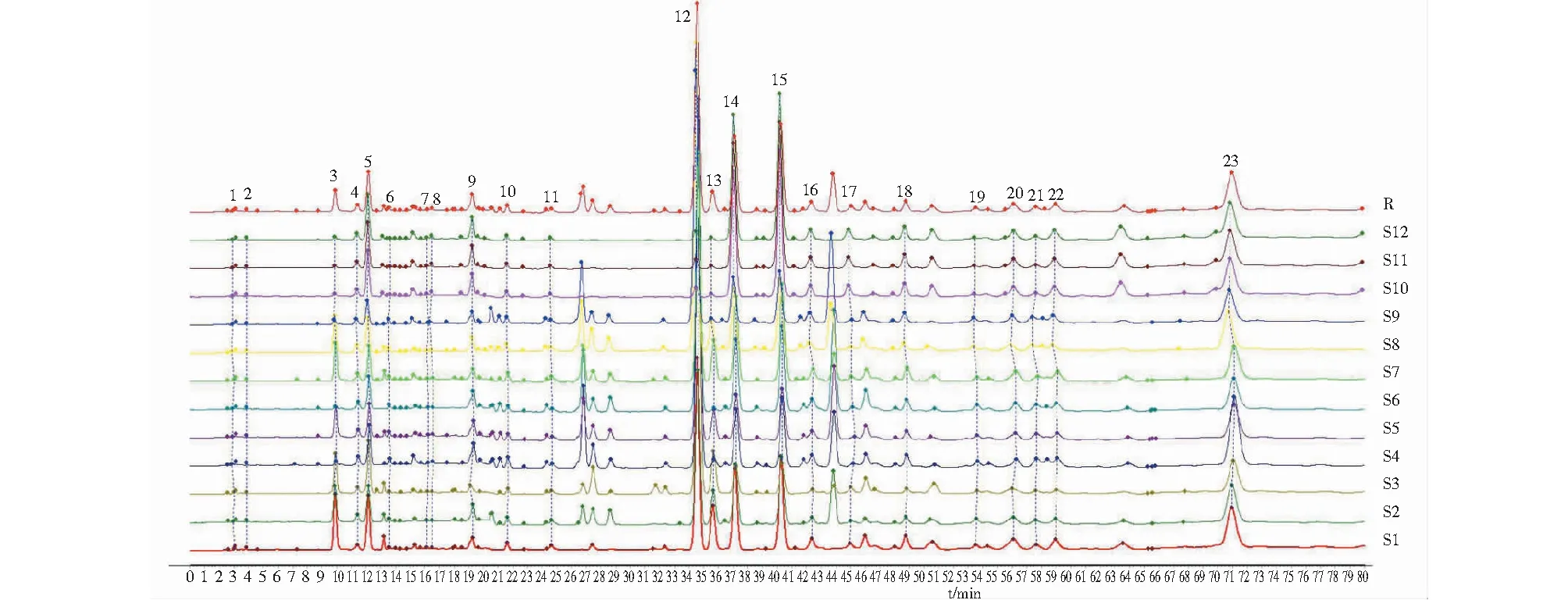
图1 12批芦荟药材的指纹图谱Fig.1 HPLC fingerprint of 12 batches of aloe

图2 芦荟样品(a)和混合对照品溶液(b)HPLC 图Fig.2 HPLC chromatograms of sample (a) of aloe and mixed reference substances (b)注:1:芦荟苦素;2:芦荟新苷D;3:7-O-甲基芦荟新甙A;4:芦荟苷B;5:芦荟苷A;6:芦荟大黄素。Note:1:Aloesin;2:Aloeresin D;3:7-O-Methylaloeresin A;4:Aloin B;5:Aloin A;6:Aloe-emodin.
2.3.1 共有峰的指认
从12批芦荟药材中标定了23个共有峰,通过与对照品比对,指认出6个成分,分别是3号峰为芦荟苦素、12号峰为芦荟新苷D、13号峰为7-O-甲基芦荟新甙A、14号峰为芦荟苷B、15号峰为芦荟苷A、23号峰为芦荟大黄素。芦荟样品和混合对照品的HPLC图谱见图2。结果表明12批芦荟药材中23个共有峰的相对保留时间RSD为0%~0.55%,相对峰面积RSD为16.54%~97.02%,表明方法稳定,但不同产地的芦荟药材中成分含量差异较大。
2.3.2 相似度分析
采用《中药色谱指纹图谱相似度评价系统(2012 A版)》软件建立12批芦荟药材共有模式的指纹图谱,其相似度评价见表1。除产地是广西的S10~S12样品相似度小于0.5外,其余样品的相似度均>0.93。
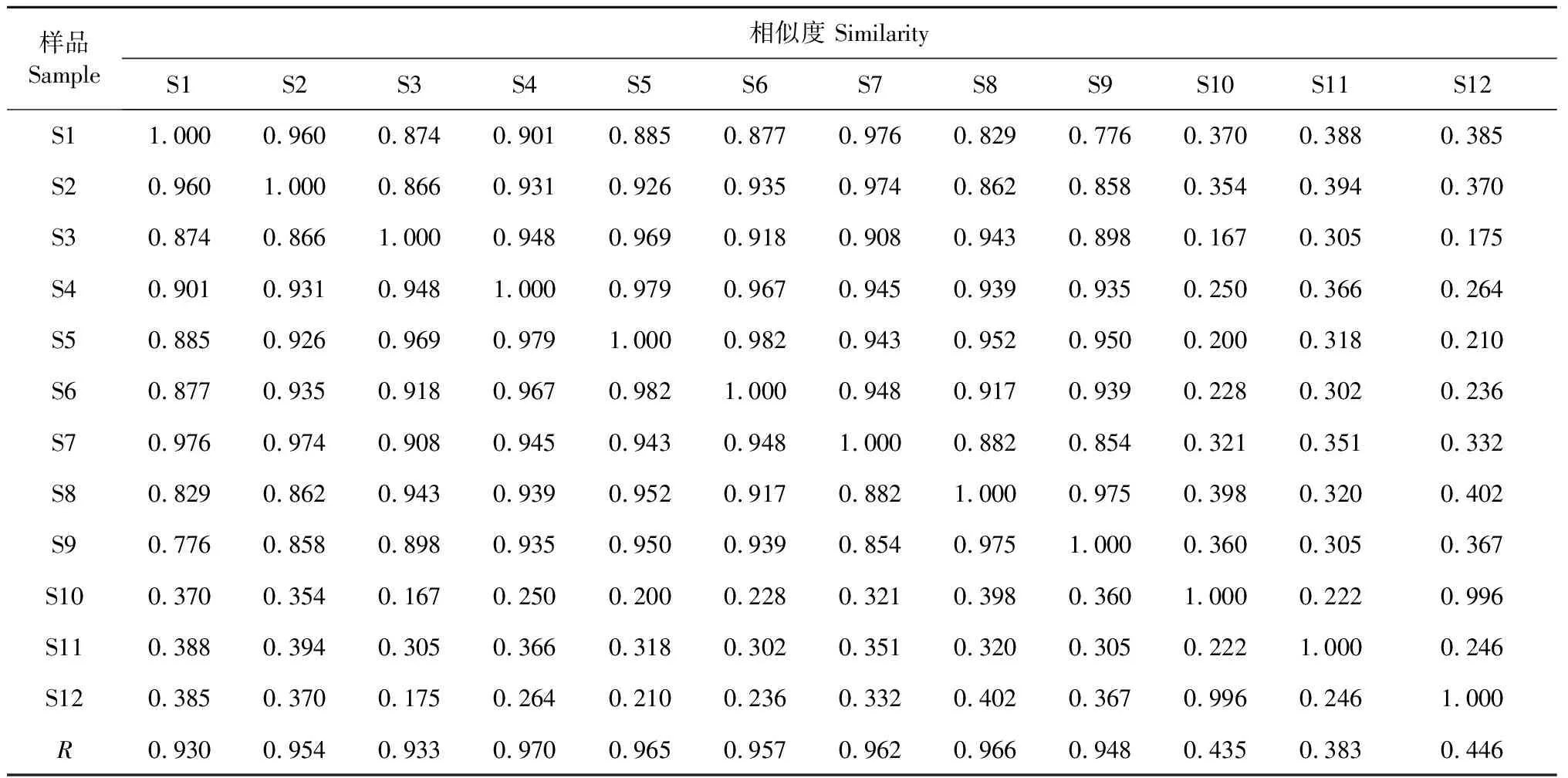
表1 12批芦荟药材指纹图谱的相似度评价表
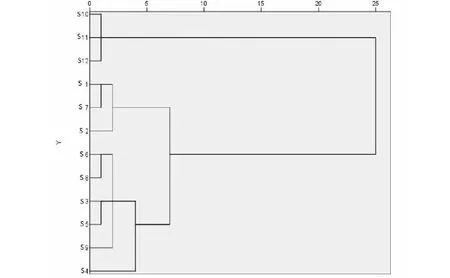
图3 12批芦荟药材的聚类分析树状图 Fig.3 Hierarchical cluster analysis plot for 12 batches of aloe
2.4 化学模式识别分析
2.4.1 聚类分析
以12批芦荟药材中23个共有峰的峰面积为变量,运用SPSS 26.0统计软件,以组间联接平方欧式距离测度的方式进行聚类分析,结果见图3。由图3可知,12批芦荟药材样品明显聚为三类,S1、S2、S7聚为第一类,S10~S12聚为第二类,S3~S6、S8、S9聚为第三类。
2.4.2 主成分分析(PCA)
采用SPSS 26.0 统计学软件对12批芦荟药材中23个共有峰的峰面积进行标准化处理,计算相关矩阵的特征值即方差贡献率,以特征值>1为标准,得到了前4个主成分的累积方差贡献率为87.593%,能够较好的代表指纹图谱中的大部分信息。从成分载荷矩阵表可以看出,第一主成分的独立方差贡献率为39.529%,主要反映色谱峰7~9、11~15、17、18、22的信息;第二主成分的独立方差贡献率为26.749%,主要反映色谱峰1、2、4、6、10、16、19、21、23的信息;第三主成分的独立方差贡献率为14.833%,主要反映色谱峰3、20的信息;第四主成分的独立方差贡献率为6.482%,主要反映色谱峰 5的信息。利用SIMCA 14.1分析软件绘制主成分得分图,见图4。结果表明12批芦荟药材大致可被分为三类,S1、S2、S7在主成分得分图的中间分为第一类;S10~S12在主成分得分图的左侧分为第二类,S3~S6、S8、S9在主成分得分图的右侧分为第三类,其结果与聚类分析结果一致,也验证了聚类分析的分类结果。
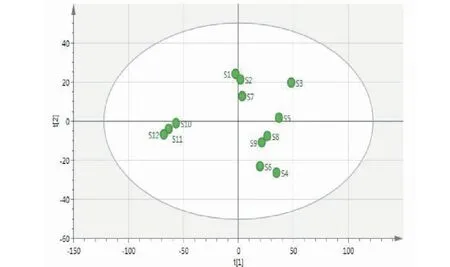
图4 主成分分析得分图Fig.4 Score scatter plot of PCA
2.4.3 正交偏最小二乘法-判别分析(OPLS-DA)
正交偏最小二乘判别分析法(OPLS-DA)是常用来处理分类和判别问题的一种非常有效的监督分析方法。而OPLS-DA模型的拟合效果评价指标主要是累积解释能力参数(R2X、R2Y)和预测能力参数(Q2),如果R2X、R2Y和Q2均大于0.5,且数值越接近于1,则表示模型拟合效果越好[17]。采用SMICA 14.1软件对12批芦荟药材共有峰进行OPLS-DA分析,拟合的模型R2X=0.929,R2Y=0.939,Q2=0.835,均大于0.8,表示拟合的预测模型稳定、可靠,可用来评价芦荟药材指纹图谱的模式识别方法,其得分散点图见图5。由图5可知,广西和四川产区的样品各自聚为一类;广东和海南产区的样品聚为一类,说明这两个产区药材的化学成分相似性很高。以变量投影重要性(VIP)值大于1为依据,筛选对分组结果产生较大影响的色谱峰,结果见图6。由图6可知,VIP值大于1的色谱峰从大到小分别为12号峰(芦荟新苷D,VIP值为2.982 3)、15号峰(芦荟苷A,VIP值为1.971 5)、13号峰(7-O-甲基芦荟新甙A,VIP值为1.691 5)、14号峰(芦荟苷B,VIP值为1.540 1)、3号峰(芦荟苦素,VIP值为1.523 5),表明这5个化合物是影响芦荟药材质量差异的标志物。

图5 OPLS-DA得分散点图Fig.5 Score scatter plot of OPLS-DA
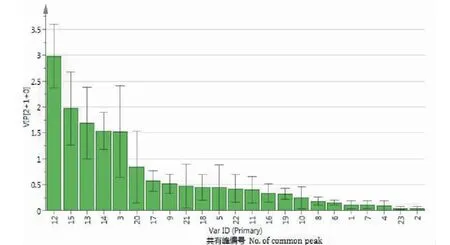
图6 12批芦荟药材OPLS-DA模型中共有峰的VIP值Fig.6 VIP values of common peaks in OPLS-DA model of 12 batches of aloe
2.5 含量测定
2.5.1 色谱条件、样品溶液制备和混合对照品溶液制备
同“2.1”一致,样品和混合对照品见图2。
2.5.2 线性关系、检测限和定量限
分别精密吸取“2.1.2”项下6个对照品贮备液适量,混合,加甲醇定容、稀释,制成一系列不同浓度的混合对照品溶液,按“2.1.3”项下色谱条件进样测定,记录峰面积。以各待测成分质量浓度(x,μg/mL)为横坐标、峰面积(Y)为纵坐标进行回归分析,结果见表2。分别精密吸取对照品溶液适量,加甲醇稀释,按“2.1.3”项下色谱条件进样测定,以信噪比10∶1、3∶1分别计算定量限、检测限。结果表明,芦荟苦素、芦荟新苷D、7-O-甲基芦荟新甙A、芦荟苷 B、芦荟苷A和芦荟大黄素的标准曲线在其各自的线性范围内均具有良好的线性关系,相关系数R2均≥0.999 5,检测限范围在0.017 4~0.314 0 μg/mL,定量限范围在0.034 9~0.559 0 μg/mL,结果见表2。
2.5.3 精密度、稳定性、重复性和回收率
精密度试验:取样品(S1),按“2.1.1”方法制备样品溶液,按“2.1.3”项下色谱条件连续进样测定6次,记录峰面积。结果显示,芦荟苦素、芦荟新苷D、7-O-甲基芦荟新甙A、芦荟苷B、芦荟苷A和芦荟大黄素峰面积的RSD分别为0.69%、0.75%、0.64%、1.22%、0.99%、1.13%(n=6),表明仪器精密度良好。
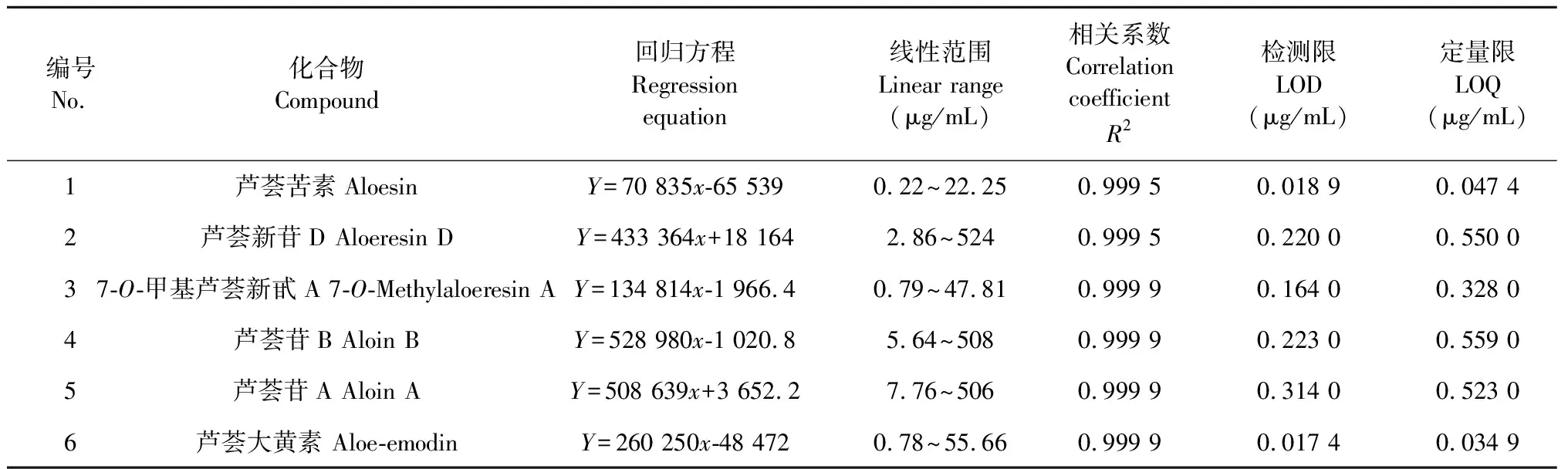
表2 6个指标成分的线性方程、相关系数、线性范围、检测限和定量限
稳定性试验:取样品(S1),按“2.1.1”方法制备样品溶液,置于室温储存,按“2.1.3”色谱条件,分别在0、2、4、8、12、18、24、48 h进样测定,记录峰面积。结果显示,芦荟苦素、芦荟新苷D、7-O-甲基芦荟新甙A、芦荟苷B、芦荟苷A和芦荟大黄素峰面积的RSD分别为1.92%、2.59%、2.73%、2.93%、2.46%、2.15%(n=8),表明芦荟样品溶液于室温下放置24 h内稳定性良好。
重复性试验:取样品(S1),精密称取6份,按“2.1.1”方法制备样品溶液,按“2.1.3”色谱条件进样测定,记录峰面积计算样品含量。结果显示,芦荟苦素、芦荟新苷D、7-O-甲基芦荟新甙A、芦荟苷B、芦荟苷A和芦荟大黄素平均含量分别为8.42、167.28、18.62、109.60、191.91、24.02 mg/g,含量的RSD分别为0.88%、0.72%、2.13%、1.30%、1.48%、1.95%(n=6),表明方法重现性良好。
加样回收率试验:取样品(S1)药材粉末(过5号筛)25 mg,按照样品中芦荟苦素、芦荟新苷D、7-O-甲基芦荟新甙A、芦荟苷B、芦荟苷A和芦荟大黄素的含有量与混合对照品比例1∶1精密加入,后续处理同“2.1.1”项。按“2.1.3”色谱条件进行测定,计算回收率。芦荟苦素、芦荟新苷D、7-O-甲基芦荟新甙A、芦荟苷B、芦荟苷A和芦荟大黄素的平均加样回收率分别为99.00%、91.45%、97.93%、94.70%、96.41%、95.34%,其RSD值分别为1.16%、0.86%、0.98%、1.13%、1.22%、1.94%(n=6)品的回收率良好,结果见表3。
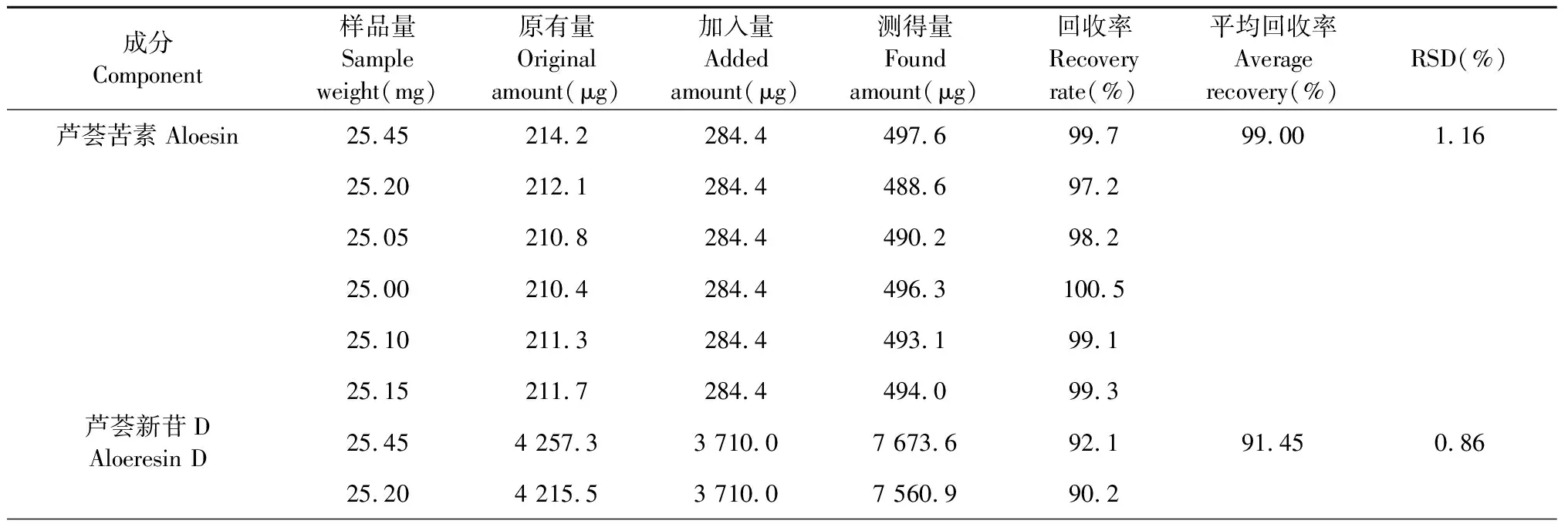
表3 加样回收率结果(n=6)
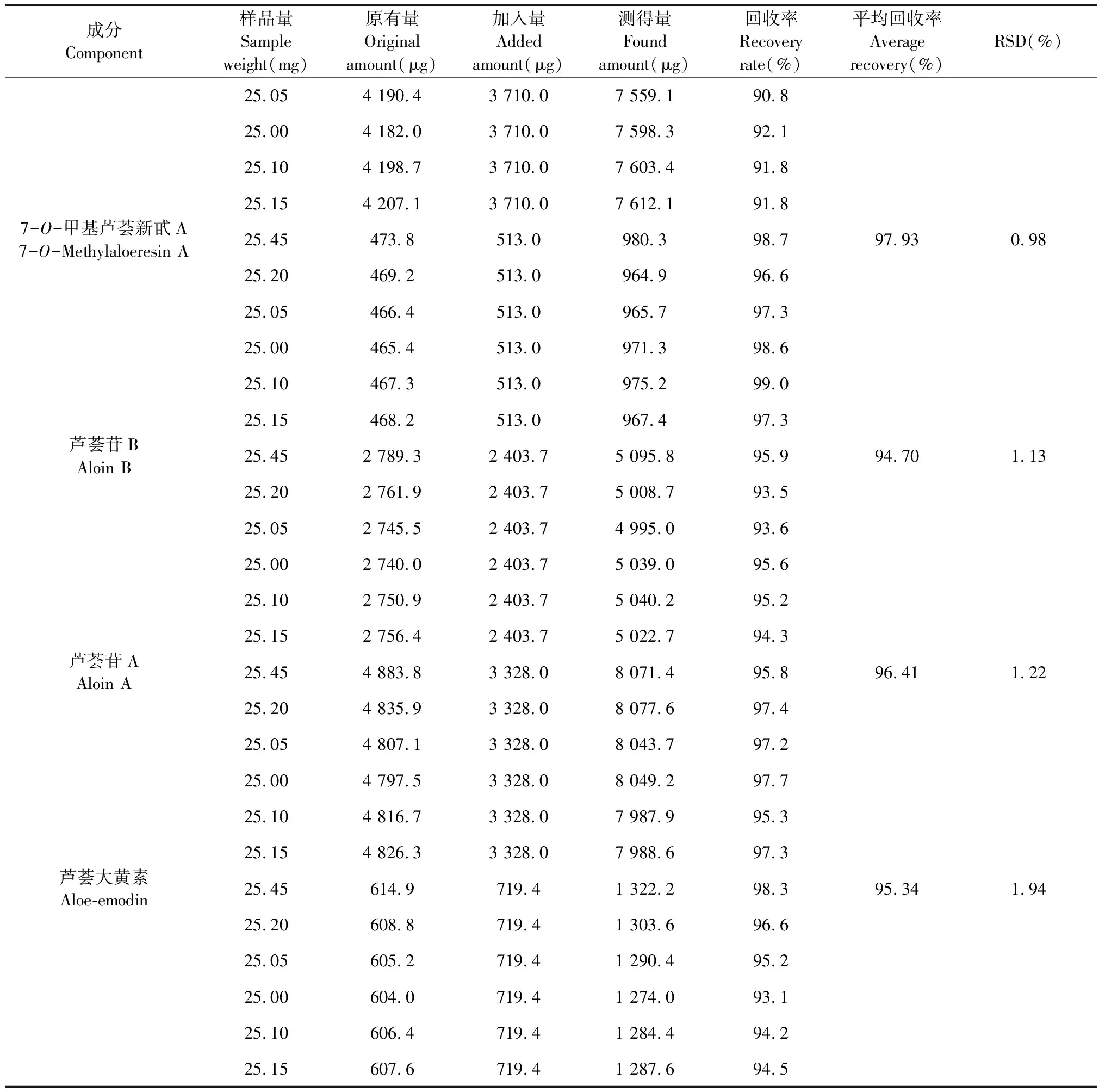
续表3
2.5.4 样品含量测定
采用“2.1.3”项下的色谱方法,测定了12批芦荟药材中芦荟苦素、芦荟新苷D、7-O-甲基芦荟新甙A、芦荟苷B、芦荟苷A和芦荟大黄素的含量,每个样品做3个平行,结果见表4。
由表4可知,广东、海南和四川产区的6个指标成分总含量达到519.87~688.87 mg/g以上,而广西产区仅达到371.31~442.73 mg/g。12批药材中6个指标成分含量对比,芦荟苦素、芦荟新苷D、7-O-甲基芦荟新甙A的相对含量差异较大,分别为0.55~13.47、7.98~369.88、1.36~25.44 mg/g;芦荟苷B、A和芦荟大黄素的含量差异相对较小,含量分别为65.14~150.08、109.51~263.16、18.30~40.32 mg/g。其中广西产区的样品,芦荟新苷D的平均含量(9.70 mg/g)远低于广东产区(293.97 mg/g)、海南产区(293.57 mg/g)和四川产区(257.81 mg/g)的平均值,但芦荟苷B(142.58 mg/g)和A(237.08 mg/g)的平均含量均高于广东产区(110.21、195.79 mg/g)、海南产区(102.80、179.87 mg/g)和四川产区(97.17、170.48mg/g)的平均值。
3 讨论
本研究比较了热回流法和超声法对特征色谱峰提取率的影响。结果发现,超声处理提取效率高、时间短、损失少,易操作。同时分别考察了提取溶剂(甲醇、乙醇)及浓度、提取时间和液料比对芦荟药材中6个指标成分提取率的影响,结果显示,50%甲醇水溶液75 mL超声提取20 min为最佳。考察了乙腈-甲酸水溶液、乙腈-磷酸水溶液和甲醇-磷酸水等流动相体系。结果表明,当以甲醇-乙腈-0.3%磷酸水溶液为流动相进行梯度洗脱时,各指标成分峰型对称、分离度较好,运行时间短,效果最佳。比较了不同检测波长(254、280、310、330、360 nm)下各色谱峰的峰形和分离度,结果显示,254 nm处6个指标成分吸收强度高,基线平稳、分离度良好,故选择254 nm为检测波长。
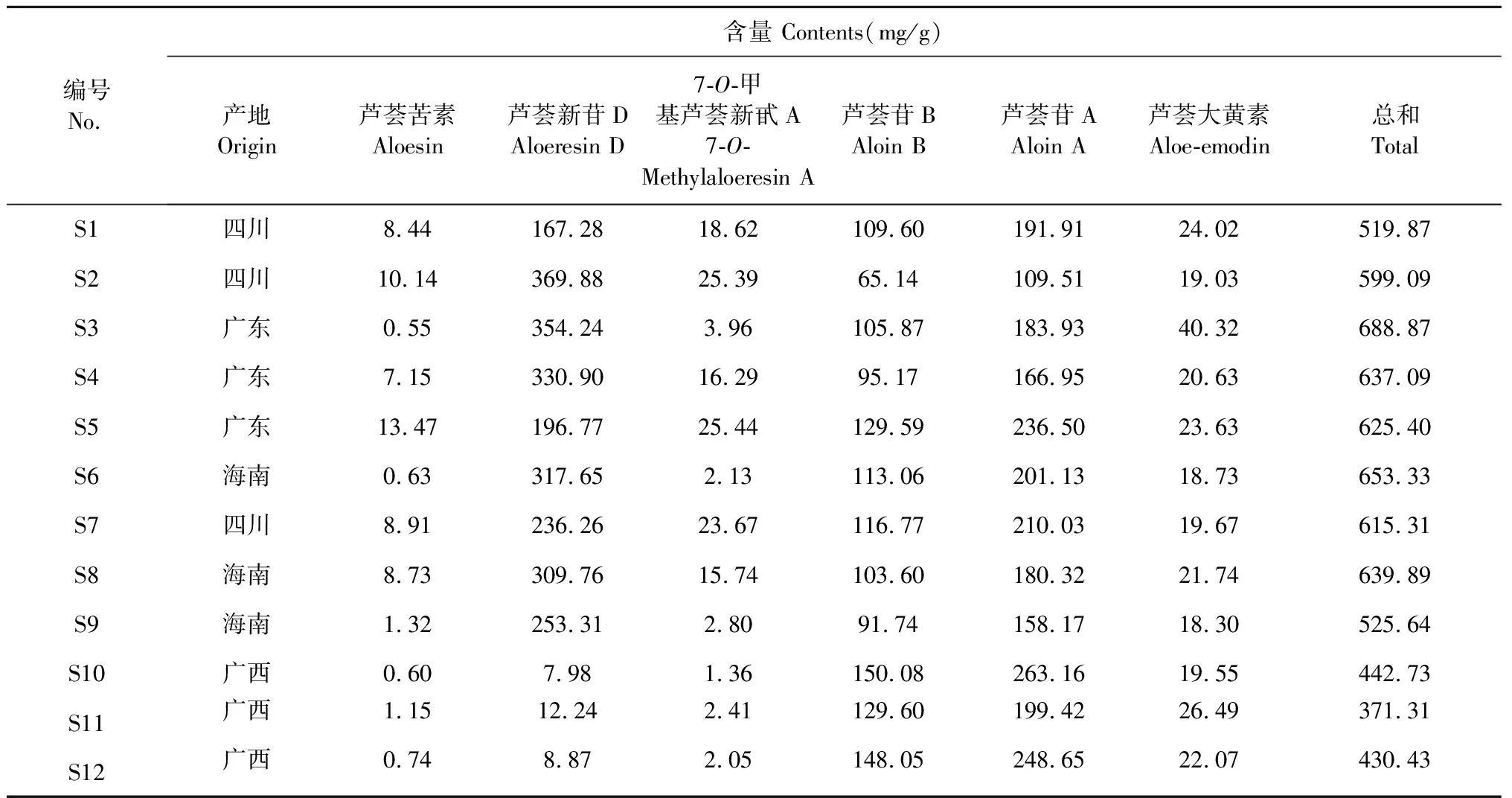
表4 12批芦荟样品6个指标成分的含量
12批芦荟药材的指纹图谱共识别23个共有峰,从相似度结果可知,除广西产区的相似度低于0.5外,其他均大于0.93,说明市售的芦荟药材因产地不同,其化学成分种类和含量有很大差异。从聚类分析结果可知,12批芦荟药材聚为3类,与主成分分析结果一致。利用正交偏最小二乘判别法筛选出影响芦荟药材质量差异的5个色谱峰,并以相同HPLC法对其进行含量测定。因芦荟大黄素也是芦荟药材中主要的生物活性化合物之一[21],固也列入含量测定的指标性成分,其分析结果可知6个指标成分含量存在较大差异。这种差异可能是由于地理环境、气候条件、采收期、土壤及田间管理、种植模式等诸多因素,对药材次生代谢产物的积累产生影响,从而导致药材相似度以及含量的差异性[22];其次芦荟药材是以浓缩物的性状呈现,其浓缩方式、烘干工艺、取材部位等也会使药材的化学成分有很大差异。
本研究采用定量指纹图谱结合化学模式多元判别方法对不同产地的芦荟药材成分差异进行分析,并对6个指标成分进行了含量测定,可直观评价各产地芦荟药材品质的一致性,还同时体现化学成分的差异性,为芦荟药材的质量控制和后续相关产品开发提供科学参考。
猜你喜欢
今日农业(2022年2期)2022-11-16
北京航空航天大学学报(2022年8期)2022-08-31
小读者(2021年6期)2021-07-22
今日农业(2021年6期)2021-06-09
作文评点报·小学三、四年级(2019年14期)2019-04-30
新城乡(2018年6期)2018-07-09
领导科学论坛(2016年9期)2016-06-05
农村百事通(2016年5期)2016-05-14
故事作文·低年级(2009年5期)2009-05-06
中国报道(2009年12期)2009-01-15
