
主要麻类作物基因组学与遗传改良: 现状与展望
2021-04-06 14:12张力岚祁建民张列梅张立武
作物学报 2021年6期
徐 益 张力岚 祁建民 张列梅 张立武,*
专题
主要麻类作物基因组学与遗传改良: 现状与展望
徐 益1,2,3张力岚1,2,3祁建民1,2张列梅1张立武1,2,3,*
1福建农林大学农学院/ 作物遗传育种与综合利用教育部重点实验室 / 福建省作物设计育种重点实验室, 福建福州 350002;2福建农林大学农业农村部东南黄红麻实验观测站 / 福建省麻类种质资源共享平台 / 福建省南方经济作物遗传育种与多用途开发国际科技合作基地, 福建福州 350002;3福建农林大学海峡联合研究院基因组与生物技术中心, 福建福州 350002
随着测序技术的发展, 主要麻类作物(黄麻、红麻、苎麻、亚麻和工业大麻)参考基因组从2011年至2020年陆续完成测序, 这标志着麻类作物科学已经进入基因组时代。文章首先详细概述主要麻类作物基因组测序。其次, 评述了基于基因组学的麻类作物重要应用价值基因挖掘。基于参考基因组和转录组测序, 大量关于纤维发育、响应非生物胁迫的候选基因被挖掘, 以促进麻类作物纤维的物种特性和“不与粮食争好地”的逆境农业。同时不同麻类作物特异性状候选基因陆续被报道, 如红麻雄性不育、亚麻种子含油量和大麻大麻素相关候选基因等。再次, 麻类作物基因组测序完成为基于组学的麻类作物遗传改良提供可能: 有助于麻类作物种质资源形成和演化机制研究, 系统解析纤维产量、纤维品质、抗病耐逆等农艺性状形成的分子基础; 有助于建立高通量基因型-表型数据库, 挖掘优异基因资源与创制新种质; 有助于创新并集成分子标记辅助选择、基因组选择、转基因等技术, 建立高效的快速育种技术体系。宜选育高产高效、抗逆抗病、适宜轻简化机械化、优质专用的多用途麻类作物新品种, 以满足麻类作物相关产业的市场需求, 适应麻类作物生产方式。尽管已经获得重要基因以及位点的信息, 但如何高效率利用已有基因资源对麻类作物进行遗传改良仍需面临一系列挑战, 如成熟稳定的遗传转化体系、麻类作物基因编辑体系构建及基因组选择育种等。
主要麻类作物; 基因组; 基因; 遗传改良
黄麻(spp.)、红麻()、苎麻(spp.)、亚麻()与工业大麻(L.)在种植历史上曾经超过300万公顷面积, 属于大面积栽培的麻类作物(以下简称主要麻类作物), 与粮、棉、油、菜并列为第五大作物群, 其种植历史可追溯到远古新石器至尧舜时代[1-2]。主要麻类作物是重要的天然韧皮部纤维作物, 其韧皮部纤维(麻皮)主要用于麻线、麻绳、麻袋、麻布等包装用纺织, 还用于家饰板材等多功能用途; 其天然活性成分在食品、日用、医药等领域价值逐步开发, 推动生命健康产业发展。主要麻类作物分布范围广, 在热带、亚热带、温带和寒带均有分布。我国的华南、长江流域、黄淮流域、淮河流域到东北(辽宁、吉林、黑龙江)、西北(新疆、宁夏、内蒙古)等均可以种植。主要麻类作物生物学产量高, 且耐盐碱、耐干旱、耐洪涝、适应性广, 不与粮、棉、油、菜争好地, 可用于盐碱地或重金属污染农田的植物修复[1], 发展主要麻类作物将带动我国逆境农业和麻类作物多功能利用的发展。
自1986年提出基因组学的概念, 其相关研究不断深入并被广泛接受, 目前基因组学已成为生命科学领域活跃且有影响的科学[3]。模式植物拟南芥[4]率先完成全基因组测序, 这是第一个完成的植物基因组序列, 随后水稻[5]也完成全基因组测序。随着测序技术的发展, 越来越多的植物进行了全基因组测序, 其中包括主要麻类作物(黄麻[6-7]、红麻[8]、苎麻[9-10]、亚麻[11-12]和工业大麻[13-14])。这些主要麻类作物的全基因组测序, 可以提供该作物全基因组的遗传信息, 尤其是对控制重要农艺性状的位点或基因的深入挖掘, 以及对调控复杂性状的分子网络的解析奠定了重要基础。
本文旨在总结主要麻类作物基因组学和遗传改良研究的最新进展, 对于引领加快麻类作物遗传育种研究具有重要意义。
1 主要麻类作物全基因组测序研究概况
以联合国粮食及农业组织的2007年至2018年数据来看, 在世界范围内, 黄麻和红麻在收获面积、产量和农业生产价值均占据麻类作物之首, 其次是亚麻、苎麻和工业大麻。其中我国黄麻和红麻的收获面积、产量和农业生产价值居世界第三(图1)。主要麻类作物基因组测序从2011年到2020年陆续公布, 分别涉及锦葵科、荨麻科、亚麻科和大麻科, 基因组最小的是圆果种黄麻336 Mb, 最大的是红麻1078 Mb (表1)。
1.1 黄麻基因组
黄麻基因组草图于2017年发表[6], 经组装发现, 长果种O-4和圆果种CVL-1基因组大小分别为445 Mb (13.04 Gb raw data, scaffold N50为3.3 Mb)和338 Mb (13.69 Gb raw data, scaffold N50为4.1 Mb), 分别有约8.4%和17.8%的数据无法覆盖, 表明该研究只是得到了黄麻基因组草图。为了获得高质量的黄麻参考基因组, 福建农林大学于2015年开展黄麻转录组[17]、基因组调研图和流式细胞仪分析。在此基础上, 以黄麻国家区域试验对照品种长果种宽叶长果和圆果种黄麻179为材料, 以三代测序技术 PacBio RS II为主体, Hi-C染色体构象捕获技术等技术辅助, 获得了长果种和圆果种黄麻染色体级别的参考基因组, 其基因组大小分别为361 Mb和336 Mb[7]。与已发表的黄麻长果种O-4和圆果种CVL-1基因组相比, 组装指标有了极大的提升。
黄麻全基因组的系统发育树显示[6], 圆果种和长果种黄麻均与可可和雷蒙德氏棉聚为一类, 表明黄麻属于锦葵科(Malvaceae)。需要指出的是, 关于黄麻属于锦葵科还是椴树科(Tiliaceae)一直以来存在争议。为了验证这一结论, 考虑到叶绿体表现为母系遗传, 其基因组被广泛应用于植物的系统发育关系分析, 整理圆果种黄麻和长果种黄麻[15]以及红麻[16]的叶绿体基因组的系统发育关系结果发现, 黄麻圆果种和长果种在亲缘关系上与棉属更近(附图1)。这进一步佐证黄麻属于锦葵科, 而不是基于植物形态学分类的椴树科。
收获面积和产量取自2009年至2018年数据平均值, 价值取自2007年至2016年数据平均值, 数据均来自联合国粮食及农业组织。
The harvesting area and production are taken from the average values from 2009 to 2018, the values are from the average data from 2007 to 2016, and the data are all from the Food and Agriculture Organization of the United Nations.
1.2 红麻基因组
红麻(, 2=36)是锦葵科木槿属的一年生常异花授粉作物, 属于短日照喜温类型。2020年福建农林大学率先绘制高质量红麻参考基因组图谱[8], 该研究以红麻优良品种福红952为材料, 采用二代加三代的测序策略, 同时结合Hi-C染色体构象捕获技术和高密度遗传图谱, 完成红麻全基因组测序和组装工作, 其基因组大小约为1078 Mb, contig N50为2.73 Mb, 共鉴定到66,004个基因。红麻与拟南芥、水稻、亚麻和棉花的蛋白编码基因比较发现, 2195个基因是红麻所特有的, 包括67个NAC和47个MYB转录因子。比较基因组学分析发现, 红麻与雷蒙德氏棉在分化后存在一次独立的全基因组复制事件(whole genome duplications, WGD), 导致红麻绝大多数基因拷贝数加倍。利用核心种质重测序数据, 群体遗传学分析显示, 红麻起源于非洲, 沿着非洲南部、非洲西部的路线传播到亚洲, 并在传播过程中发生了2次驯化事件。在参考基因组基础上, 该研究确定了控制红麻叶形的基因为LATE MERISTEM IDENTITY 1 (LMI1)转录因子, 并用病毒诱导的基因沉默(virus-induced gene silencing, VIGS)验证了其基因功能。
1.3 苎麻基因组
苎麻(, 2=28)为荨麻科苎麻属多年生宿根性草本植物, 单性花, 雌雄同株, 花序复穗状, 雄花花序生在茎的中下部, 雌花花序生于梢部。我国的苎麻产量占全世界苎麻产量的90%以上。
2018年2个苎麻基因组草图陆续公布[9-10], 以Zhongzhu 1为测序品种, 得到一个341.9 Mb的基因组草图, contig N50和scaffold N50分别为22.62 kb和1226.36 kb, 鉴定出30,237个蛋白编码基因。苎麻作为荨麻科中第一个被测序的物种, 可对该物种的多样性和物种形成等研究提供参考。比较基因组学发现, 苎麻有1075个独有的基因家族, 包含4082个基因, 其中分别有5个和10个被注释为假定的CesA和WAT1相关蛋白。分析这15个基因所编码的蛋白结构域发现, 5个假定CesA蛋白和3个假定WAT1蛋白有相应的保守结构域, 表明这些基因可能分别是真实的和相关基因, 且与苎麻纤维产量有关。与其他4个物种(川桑、大枣、西瓜和亚麻)的CesA和WAT1相关蛋白的系统发育分析进一步证实, 5个和3个相关基因是苎麻特有的。进化树分析发现106个自然选择的基因, 其中22个与氮代谢有关, 这些基因可能与苎麻的粗蛋白含量和营养生长性状驯化相关。这些研究为苎麻遗传育种和分子研究提供了理论基础。
1.4 亚麻基因组
2012年以CDC Bethune为测序品种, 得到一个373 Mb的基因组草图, contig N50为0.02 Mb, scaffold N50为0.69 Mb, 鉴定出43,484个基因[11]。最近, 甘肃省农业科学院分别对油用亚麻品种、纤维用亚麻品种和白胡麻测序得到染色体水平参考基因组[12], 组装得到的基因组大小为别为306.0、303.7和293.5 Mb, 其中contig N50/scaffold N50分别为131 kb/ 1235 kb、156 kb/700 kb和59 kb/384 kb, 在每个基因组中大约预测到43,500个编码基因和2600~2800个非编码RNA; 系统发育分析表明, 栽培亚麻和白胡麻在大约232万年前发生了分化, 自古被子植物的六倍化事件以来, 亚麻发生了2次WGD事件。在最近一次WGD事件中,基因拷贝数发生了扩张, 可能促成了亚麻独特的次生细胞壁生物合成, 对基因的定向选择可能形成了当前的油用和纤维用亚麻的形态类型。对83份亚麻种质资源重测序, 在油用亚麻中, 油脂相关基因(、、和)和种子大小相关基因(和)受到选择; 在纤维用亚麻中, 次生细胞壁生物合成相关基因(、和)和植物株高相关基因(、和)受到选择。
1.5 工业大麻基因组
2011年工业大麻基因组草图发表[13], 该研究以一种广泛应用于药用的大麻品种Purple Kush为测序品种, 组装得到一个534 Mb的基因组草图。2020年5月公安部物证鉴定中心发表了一个高质量染色体水平的野生大麻参考基因组[14], 其基因组大小为808 Mb, contig N50为513.57 kb, scaffold N50为83 Mb, 相对于2011年发表的大麻基因组草图(contig N50为2.8 kb; scaffold N50为16.2 kb)有很大的提升。统计其重复序列高达74.75%, 注释到了38,828个蛋白编码基因。然而目前为止籽用大麻基因组还未公布, 待籽用大麻基因组公布之后, 以期比较基因组学在3种不同用途的大麻之间挖掘更丰富的变异位点, 为不同用途的大麻育种提供更多信息。
2 主要麻类作物重要应用价值基因挖掘
纤维发育与非生物胁迫相关基因的挖掘一直是主要麻类作物遗传改良的重要方向。随着主要麻类作物全基因组测序陆续被报道, 很有必要系统总结回顾NGS (next generation sequencing)技术对主要麻类作物纤维发育相关基因挖掘(附表1)、响应各种非生物胁迫的遗传机制(附表2)和麻类作物特异性状候选基因的最新报道(附表3)。为了表述方便, 不同麻类作物分开阐述。每个麻类作物分别从纤维发育、非生物胁迫和麻类作物特异性状3个方面展开。
2.1 黄麻重要应用价值基因挖掘
2.1.1 黄麻纤维发育相关基因 纤维发育的分子机制解析属于麻类作物遗传改良研究的重要方向。为鉴定纤维发育相关的基因, 在黄麻纤维发育关键时期进行转录组测序[17], 共组装出48,914个unigenes, 其中有5个、3个、9个、18个、2个(Korrigan), 它们可能参与黄麻纤维素生物合成。长果种黄麻茎皮转录组[18]分析显示, 次生代谢产物的生物合成在代谢途径中显著富集, 其中26个基因持续上调, 包括4个参与葡聚糖代谢的基因, 调控茎皮韧皮部的发育。对一个纤维缺陷突变体进行转录组测序[19]发现,在突变体的韧皮部组织中的任何生长阶段都显著下调, 且在生长早期的表达下调是伴随着次生细胞壁特异性基因纤维素合成酶A7 ()和阿拉伯半乳聚糖糖蛋白6 ()的共同上调, 这可能与黄麻纤维中协调木聚糖S型的沉积有关。黄麻全基因组测序显示[6], 在纤维素合成途径中、、和的表达较高。抑制差减杂交[20]被成功应用于黄麻纤维发育过程中相关基因的鉴定, 以纤维素含量极低的突变体作为试验组, 纤维素含量正常的品种作为对照组, 共鉴定出377个功能已知的基因。其中,、和在正常植株中的表达明显高于突变体, 表明这些基因可能参与黄麻木质素合成, 且是起主要作用。
2.1.2 黄麻非生物胁迫响应基因 解析麻类作物非生物胁迫响应的分子机制是其遗传改良的又一重要方向, 包括盐胁迫和干旱胁迫等。黄麻盐胁迫下转录组测序[21]显示, 在叶和根中分别检测到32个和196个差异表达转录因子, 其中有11个在2个组织中均被检测到; 在叶片中的差异表达转录因子家族包括HB和HSF, 而在根中的差异表达转录因子家族包括MYB、WRKY和CCAAT。脱落酸ABA信号通路是植物盐胁迫反应的核心, 在此途径上发现了20个差异表达基因, 包括编码PP2C、SnRK2、ABF (上调)和PYL (下调)的基因。在细胞分裂素途径上发现, 在高浓度盐胁迫下, 叶和根中的、和均下调, 而大多数基因上调, 表明它们负调控细胞分裂素信号以提高植物的耐盐性。黄麻耐旱品种和敏感品种的比较转录组分析[22], 在干旱敏感型品种中, 鉴定出34个转录因子差异表达基因和23个蛋白激酶差异表达基因, 其中包括19个类受体激酶(receptor like kinases, RLKs)。这些RLKs在干旱胁迫下大部分下调, 说明它们是黄麻抗旱机制的负调控因子。此外, 3-ketoacyl-CoA合酶(3-ketoacyl-CoA synthase, KCS)的表达能显著提高黄麻抗旱性, 构建KCS基因的过表达和基因编辑载体, 并用于黄麻植株的转化, 发现KCS基因的过表达不仅增加了叶绿体的数量, 还增强了植株的抗旱性, 提高了木质素含量、生长速率、茎皮厚度和茎粗[23]。
2.1.3 黄麻特异性状相关基因 黄麻2个栽培种(或亚种)在表型上存在较大差异, 如长果种的果荚为长条形状, 种子颜色为墨绿色; 圆果种的果荚为球形, 种子颜色为棕色等。不仅如此, 长果种为常异花授粉作物, 圆果种为自花授粉作物, 然而造成2个栽培种间形态或重要性状差异的遗传机制目前还有待深入研究。
2.2 红麻重要应用价值基因挖掘
2.2.1 红麻纤维发育相关基因 与黄麻纤维不同的是红麻韧皮纤维中纤维素含量相对较低, 木质素含量较高。红麻基因组[8]不仅揭示了一些参与韧皮纤维次生壁合成和纤维素沉积的基因, 如:、和等, 还结合QTL定位鉴定出和这2个可能参与细胞壁形成的基因, 而也是红麻在驯化过程中的受选择基因。因此可能在红麻纤维发育过程中发挥重要作用。转录组分析揭示出317个淀粉和糖代谢途径的基因, 可能参与纤维素生物合成[24]。在此之前、、、、和被报道在木质素生物合成中起关键作用[25]。红麻在开花后纤维累积速度显著降低, 因此延后红麻开花期也是提高纤维产量的一个途径, 在红麻光周期调控途径中可能存在类似拟南芥GI-CO-FT的保守途径: 短日照条件下,表达量上调促进表达, 从而促进植物开花,和这2个基因在光敏感和光钝感种质中表达存在差异[26], 说明这2个基因可能参与红麻光周期调控。
2.2.2 红麻非生物胁迫响应基因 红麻本身作为一种耐旱、耐盐碱、耐贫瘠、易栽培的作物[1], 研究红麻耐盐碱机制, 培育红麻耐盐碱种质可将红麻种植转向盐碱地, 减少用地压力。比较盐胁迫下红麻的转录谱[27]发现, 盐胁迫下转录因子NAC、BZIP、AP2/EREBP、ARF、AP2/ERF、bHLH、TCP均上调, 有8个NAC转录因子和2个WRKY转录因子表达下调, 这些转录因子可能通过多种不同途径参与了红麻的耐盐性。基于转录组, 不少学者对红麻非生物胁迫产生响应的基因进入了深入挖掘, 认为基因是红麻ABA和MeJA信号转导途径、盐和干旱胁迫应答途径的关键枢纽基因[28]; 外源镉和盐处理能够显著诱导表达[29], 该基因可能参与调控红麻逆境胁迫反应, 但仍需要更多试验验证在红麻逆境胁迫中的生物学功能。Wei等[30]分离鉴定红麻6个HDACs基因(、、、), 亚细胞定位显示,和位于细胞核中,和位于细胞核和胞质中, 而位于细胞核和质膜中, 这6个基因均为红麻盐胁迫和干旱胁迫的响应基因。
2.2.3 红麻特异性状相关基因 作物细胞质雄性不育(cytoplasmic male sterile, CMS)是作物杂种优势利用的主要途径, 2004年选育出红麻CMS系K03A[31], 实现了三系配套, 自此红麻育性相关研究越来越多, 与红麻育性相关的基因也被陆续报道。赵艳红等[32]在红麻线粒体基因中发现一个与雄性不育细胞质相关的47 bp缺失, 这47 bp缺失片段分别与线粒体转运信号和GFP的ATP9融合, 形成2个嵌合基因和,将含嵌合基因的载体转入烟草发现, 部分转基因植株为雄性不育后半不育[33], 并基于基因开发的分子标签MM556可用于红麻不育细胞质的鉴定[34]。对不育系和在保持系花药进行比较转录组分析[35]发现,在三羧酸循环(tricarboxylic acid cycle, TCA cycle)中起直接作用, 在不育系中表达量下降了5.6倍。随后基因也被报道因为其CDS中存在33-bp的缺失和3-bp的插入, 导致红麻细胞质雄性不育[36]。ATP6与MYC2b蛋白之间存在互作, 导致红麻细胞质雄性不育, 而ATP6与MYC2a、MYC2b蛋白之间均存在互作时, 红麻表现为可育[37]。基因编码一个富含亮氨酸重复的F-box蛋白, 而F-box蛋白被报道出在花器官发育中有重要功能[38], 且在花药期不育系中基因表达显著高于保持系, 这可能导致花药败育[39]。目前认为雄性不育主要包括由线粒体基因和核基因共同控制, 然而又有报道通过RNA编辑技术发现叶绿体基因也可能与红麻细胞质雄性不育有关[40]。红麻细胞质雄性不育基因及其调控机制还有待深入研究。
2.3 苎麻重要应用价值基因挖掘
2.3.1 苎麻纤维发育相关基因 苎麻纤维中纤维素含量高于黄麻且木质素含量远低于黄麻, 纤维品质比黄麻好。转录组测序[41]鉴定出36个纤维素合酶基因, 其中有33个在茎皮中的表达远高于其他组织。随后[42]、、[43]和[44]也都被陆续报道在韧皮部或木质部表达参与次生细胞壁合成。对苎麻野生种和栽培种的转录组进行比较[45]发现, 高纤维素含量品种的驯化可能与基因的正向选择有关。相关的基因在苎麻基因组[10]中也被注释为与纤维产量的相关基因。对木质素含量的遗传基础进行分析发现[46], 3个位于木质素含量QTL区域的基因, 在纤维发育的2个不同阶段都有不同的表达, 且它们分别编码MYB蛋白、莽草酸羟基肉桂酰转移酶和漆酶。Tang等[47]发现,、、、、和的表达量均与木质素含量成正比, 但苎麻4CL3重组蛋白在催化过程中以肉桂酸为底物, 因此对木质素合成有负调控作用, 推测苎麻主要参与苎麻黄酮类化合物的合成,主要参与木质素的合成。探索与对调控苎麻纤维次生细胞壁发育的作用发现[48],和过表达能够上调苎麻次生壁发育关键基因的表达量, 从而导致次生壁纤维的生物合成增加;有可能直接参与细胞程序性死亡调控次生细胞壁发育, 而可能在苎麻纤维成熟中发挥重要作用。对不同区段的茎皮进行转录组测序[49]发现, 乙烯和赤霉素可能参与的苎麻韧皮纤维组织发育, 这说明研究调控苎麻纤维发育的相关基因可以间接从乙烯和赤霉素合成酶入手。
2.3.2 苎麻非生物胁迫响应基因 非生物胁迫严重影响苎麻的产量与品质。基于苎麻转录组, 筛选出8个WRKY转录因子, 与其他作物已报道的10个具有抗旱功能的WRKY转录因子对比, 推断这个8个WRKY转录因子也可能参与苎麻干旱胁迫响应[50]。[51]、[52]和[53]也被证实响应苎麻ABA、干旱和高盐逆境胁迫, 将苎麻基因转入烟草发现, 其显著提高转基因植株的生物量和氮利用效率[54], 从而减轻外界非生物胁迫。[55]、[56]、[57]和[58]也都被陆续报道出能响应重金属镉胁迫。此外, Chen等[59]发现73个miRNA, 它们介导苎麻对重金属镉胁迫的响应。
2.3.3 苎麻特异性状相关基因 苎麻是一个杂种优势利用率较高的作物, 对苎麻育性相关基因的挖掘能为其雄性不育机理研究奠定基础。目前已经报道出的与苎麻育性相关的基因有[60]、和[61]。
2.4 亚麻重要应用价值基因挖掘
2.4.1 亚麻纤维发育相关基因 亚麻纤维中纤维素含量高于黄麻, 木质素含量远低于黄麻, 纤维品质优于黄麻。基于亚麻基因组信息, 鉴定出32个与亚麻纤维发育相关的候选基因, 其中16个编码纤维素合成酶(CesA), 16个编码纤维素合成酶类蛋白(Csl)[62], 利用VIGS沉默这些基因后, 韧皮纤维的数量和结构受到了严重的影响[63]。亚麻纤维素生物合成的调节涉及的表达调控、胞质中纤维素合酶复合体的装配、纤维素的沉积等多个方面, 但对Csl蛋白的功能尚不清楚[64], 而基因的转录水平主要依赖于产生的蛋白质的功能亚类和植物发育阶段[65]。对亚麻不同茎段的转录组分析显示[66], 在次生细胞壁形成的晚期韧皮纤维中(茎底部区域), 编码金属硫蛋白、脂质转移蛋白以及参与蛋白质合成/翻译和泛素介导降解的候选基因表达量增加; 与顶杆区相比, 分化后期的韧皮纤维中有156个基因上调, 其中有与细胞壁相关的基因; 与其他茎段相比, 参与次生细胞壁沉积的转录因子在韧皮纤维中的表达减少。除以上基因家族外,基因主要参与S-木质素的生物合成, 转基因植物在活性被抑制时, 木质素含量减少[67];基因表达量与亚麻纤维拉力强度成正比[68]; Ca2+信号通路通过对基因的转录调控, 在木质素生物合成的ABA调控中起着至关重要的作用, 而是该调控的关键调节因子[68];通过与启动子中的W-box结合, 在响应ABA和尖孢镰刀菌诱导的木质素生物合成中发挥调节作用[69]。
2.4.2 亚麻非生物胁迫响应基因 亚麻基因组测序完成为全基因组水平分析非生物胁迫响应基因提供了前提条件。在亚麻全基因组中共找到137个类抗病基因[70], 34个基因响应高温胁迫[71]。转录组测序发现[72],和基因家族在亚麻对土壤营养胁迫的响应中起着重要的作用,基因产物可能通过Ca2+介导的胞内调节参与了亚麻对高酸性、高Al3+浓度的响应[73], MADS-box和NAC转录因子参与调节植物生长发育和参与亚麻耐铝性细胞壁修饰的酶[74]。干旱胁迫对亚麻产量和油质量的影响主要表现在生育期, 选择性扫描发现了与脱落酸途径、生长素信号、Ca2+信号、光合作用调节和干旱应答转录因子有关的各种基因[75]。亚麻锈病感染转录组[76]揭示6个基因(、、、、()和)参与了亚麻抵御锈病感染, 在感染早期表达量达到一个峰值, 随着孢子形成逐渐下降。
2.4.3 亚麻特异性状相关基因 籽用亚麻种子含油量一般为30%~45%, 早在史前时代就被作为油料作物种植。对亚麻未成熟胚中磷脂酶基因家族的转录组学分析[77]发现, PLA2在亚麻中主要参与甘油磷脂代谢途径和醚酯代谢途径; PLC除参与甘油磷脂代谢和醚酯代谢之外, 还在肌醇磷酸代谢途径中发挥重要作用。在TAG合成途径中发现7个关键基因, 其中、和基因的动态表达模式与含油量的动态积累模式显著正相关,的动态表达模式与亚麻酸的动态积累模式显著正相关, 且在高油高亚麻酸材料的种子动态发育阶段中基因的累积表达量也显著高于低油低亚麻酸材料, 因此可能是影响籽用亚麻(胡麻)不同品种(系)中含油量和亚麻酸含量的关键基因[78]。全基因组关联分析和结合转录组测序[79], 筛选出10个候选基因, 其中6个基因参与了重要的脂肪酸代谢途径, 其中一些基因还具有上下游调节关系。对不同亚麻酸含量的亚麻品种进行不同时期的品质测定发现,、和这3个基因参与不饱和脂肪酸积累过程。
2.5 工业大麻重要应用价值基因挖掘
2.5.1 工业大麻纤维发育相关基因 工业大麻是一种多用途作物, 韧皮纤维主要用于纺织和生物复合工业。其下胚轴有一个主动伸长, 然后变厚形成初生和次生韧皮纤维的阶段, 因此工业大麻是研究二次生长过程的合适模型[80]。工业大麻下胚轴的二次生长与转录因子NST1、MYB46和WLIM1的上调有关[80-83]。韧皮纤维的伸长依赖于XTH的活性, 如和, 这些基因在伸长的大麻下胚轴中表达较多[80]。家族中参与纤维素生物合成的2个基因和, 以及次生细胞壁纤维素合成酶基因(、和)在二次生长的下胚轴中均有较高表达量[80]。和在工业大麻二次生长的下胚轴中表达量高, 此时初生和次生韧皮纤维都存在[80]。已知基因家族会影响植物细胞壁中纤维素、阿拉伯糖和半乳糖的含量, 因此也可能有助于确定韧皮纤维细胞壁的组成[84]。RNA-Seq分析发现[80], 在下胚轴二次生长过程中涉及到葡萄糖醛酸苷生物合成的几个基因的上调, 特别是涉及到主干延伸(、、和)、主干乙酰化(、)和取代基甲基化()有关的基因。
2.5.2 工业大麻特异性状相关基因 工业大麻虽然是一种纤维作物, 但大麻素作为工业大麻所特有的一类次生代谢产物, 在抗肿瘤、神经系统保护、免疫调节和抗炎抗氧化等方面具有重要的药用价值[85], 因此工业大麻作为药用植物也有很大的前景。工业大麻全基因组测序[13]鉴定出2个参与调控大麻素合成途径的基因和, 此外还鉴定出23个编码大麻素原酸(CBCA)形成的候选基因, 其中4个基因命名为~。在MEP和GPP途径后端的酶基因、、、和(lsu)表达量和大麻素含量呈现显著正相关, 在大麻素合成途径中、和3个大麻中特有的酶基因主要在始果期苞片腺毛中对大麻素合成积累起着关键作用[86]。不同工业大麻品种的腺毛转录组分析, 鉴定出之前从未报道过的编码活性酶的基因,和编码橙花醇/芳樟醇合成酶,编码大根香叶烯B合酶和编码四甲基环癸二烯甲醇合酶[87]。大麻聚酮合酶()是催化大麻素生物合成的第一步, 有研究发现, 啤酒花衍生的转录因子和番茄浓密矮化病毒(TBSV)p19共同参与了基因启动子的激活[88]。Laverty等[89]利用药用大麻和毒品大麻杂交群体构建遗传图谱发现, 大麻素的生物合成基因是不连锁的, 但产生THCA和CBDA合成酶底物的芳香族丙烯酰胺转移酶(AP)与已知的大麻素含量标记紧密连锁, 这为在大麻中如何产生大麻素提供了不同见解。
大麻作为雌雄异株植物, 性染色体进化是其又一特异性状。Prentout等[90]在大麻中发现了超过500个性连锁基因, 将这些性连锁基因定位到大麻基因组组合中, 其中有363个在1号染色体, 并且发现有1对染色体具有较大的非重组区, 进一步分析发现, 大麻具有一个强烈退化的Y染色体, 是迄今为止最古老的植物性染色体系统, 这有助于工业大麻性染色体鉴定和性别基因定位。
长期不活动将导致骨骼肌的丢失、体弱、酸血症、胰岛素抵抗及血栓形成等并发症,并将导致工作能力的下降。早期活动可以减少胸部并发症及减少不活动引起的胰岛素抵抗;联合早期下床活动及营养支持,将改善肌肉强度。研究发现,术后早期活动与ERAS的成功与否显著相关[2]。相反,术后第1天不能早期下床活动,可能是由于镇痛不足、持续的静脉输液、留置盆腔引流管、患者的动力及合并疾病等因素所导致。有研究发现,不能下床活动是影响ERAS依从性及延长住院时间重要因素之一。
3 基于基因组学的麻类作物遗传改良
3.1 我国麻类作物品种改良历程
主要麻类作物在我国均拥有悠久的种植历史, 其品种改良大致经历了以下4个阶段: 第1阶段是地方种的搜集鉴定及引种利用。在此阶段不仅大量丰富了种质资源类型, 而且鉴定出一批具有优良性状的材料, 如黄麻品种D154和翠绿等。第2阶段是杂交育种或回交选育等常规育种方法选育优良种质。此阶段是利用第1阶段所得到的优异种质为基础材料进行育种工作, 在此阶段利用杂交与诱变育种相结合, 选育出一大批高纤维产量和高含油量的亚麻品种, 如黑亚2号和亚宁1号等。第3阶段为高产、优质、多抗育种阶段。在此之前由于长期使用少数几个性状优异的骨干材料作为亲本, 使麻类作物品种遗传基础狭窄, 导致作物平均单产和品质的降低以及病虫害的发生, 如: 亚麻枯萎病和红麻炭疽病, 因此育种家们在此阶段选育出一批高产、优质、多抗育种的新品种。第4阶段为优质高产专用新品种选育和现代生物技术辅助育种阶段。由于常规育种在进一步提高产量、品质和抗性等方面的局限性逐步显现, 且很难满足市场需求的优质专用种质培育, 与此同时现代生物技术快速发展, 利用生物技术辅助育种越来越受到重视, 因此麻类作物已经由单一的纤维用新品种选育向多用途、专用型新品种选育发展, 如亚麻轮选1号和天亚7号等, 红麻福红2号和福红992等。可见, 我国主要麻类作物的育种方法正在从常规育种转变为常规育种结合现代生物技术辅助育种方法, 而育种目标也在从高产转变为优质抗病高产并重, 逐步过渡到为了适用多用途而进行专用型品种的选育。其遗传改良已经从一个完全基于表型的过程, 在某种程度上转变为越来越依赖基于基因型的选择。这得益于基因组学革命极大地提高了我们对作物基因组的理解, 使得现代育种家可以更加关注一个新的时代, 即基于基因组测序的作物遗传改良。
3.2 麻类作物育种技术的变迁
我国麻类作物育种工作开始于农家品种选育及外来优异种质引进结合常规育种手段, 如杂交育种、诱变育种等, 选育出性状优良种质。随着生物技术的高速发展, 常规育种手段结合现代生物技术的育种方法成了麻类作物的主流育种技术。较常见的3种基于生物技术的育种方法有, 一是分子标记辅助育种 (molecular marker assisted breeding, MAS)[91], 目前常用的分子标记有简单重复序列(simple sequence repeat, SSR)和单核苷酸多态性(single nucleotide polymorphism, SNP)等。这些分子标记在麻类作物中主要应用于物种演化及亲缘关系研究[92]、群体多样性研究[93]、标记辅助选择[94]和遗传图谱构建[95]等。
另一种是利用全基因组选择(genomic selection, GS)[96-97], 即利用所有可用的标记进行育种值的预测。在GS中, 所有的位点、单倍型和标记效应都是在整个基因组水平上进行估计的, 根据已知的表型和基因型数据, 以计算代表性群体中每个个体的基因组估计育种值(genome estimation breeding values, GEBVs)。连锁不平衡(linkage disequilibrium, LD)在GS方法使用中是一个非常重要的参数[97], 对于LD衰减速率快的物种, 利用没有亲缘关系的自然群体, 可以使用数量较多的标记可以在不降低检测能力的前提下筛选出用于杂交育种的亲本选择。目前全基因选择育种已经广泛应用于模式作物育种, 玉米[98]和水稻[99]等粮食作物育种中有了较深入的研究, 但在麻类作物上还鲜有报道。全基因组选择育种研究在麻类作物遗传改良中正处于起步阶段, 随着基因型分析成本的降低和统计方法的发展, 基因组选择将会逐步完善, 加快我国麻类作物育种发展的步伐[100]。
还有一种是利用基因编辑育种。随着科学技术的进步, 被称为上帝之手的基因编辑应用于遗传改良, 已成为当今的作物遗传改良研究热点。序列特异性核酸酶, 包括锌指核酸酶(zinc-finger nuclease, ZFN)和转录激活物样效应核酸酶(transcription activator-like effector nuclease, TALEN)已被初步应用于真核生物中进行靶向基因组编辑[101]。另一种基于双链断裂的基因组编辑技术CRISPR/Cas9系统, 是在细菌和古菌群中利用规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats, CRISPR)自适应免疫系统的基础上发展起来的[101], 它与ZFN和TALEN相比, 操作更简单、精确度和效率也都更高。这种新技术目前已在麻类作物上有所应用, 如构建针对基因的敲除载体[102], 为进一步实现在亚麻植株体内对基因高效率的定点编辑, 以及CRISPR/Cas9 系统在亚麻基因组编辑中的应用提供参考。
3.3 下一代测序(NGS)技术对麻类作物遗传改良带来新的策略提供可能
在下一代测序(next generation sequencing, NGS)技术之前, 一个标准的遗传连锁图谱是基于几百个分子标记来构建[103]。NGS技术的发展与许多其他新技术相结合, 使得DNA测序具有高通量和成本低的特点[104], 麻类作物基因组测序完成为基于自然变异的正向遗传学策略提供可能。目前SNP标记在主要麻类作物遗传育种研究中的应用鲜有报道, 首个黄麻SNP连锁图谱用于对RIL群体是否抗茎腐病进行基因分型, 准确率高达91%[94]。这表明, NGS技术对麻类作物高密度遗传图谱构建和重要性状定位以及标记辅助育种带来新的策略。
随着NGS技术的发展, 我们可以对麻类作物的不同品种进行深度重测序和全基因组从头测序并进行等位基因变异分析, 通过序列比对找到了大量与重要性状相关的SNP位点, 为品质改良和新品种选育带来了新的科研方法, 加快了新品种的育种进程。在没有参考基因组之前, 基于酶切的简化基因组测序(restriction-site associated DNA sequencing, RAD-seq)[105]和基因分型测序(genotyping by sequencing, GBS)[106]被应用于植物中, 简化基因组测序技术具有不依赖于基因组序列, 而进行高通量的SNP标记开发的优点。为了解亚麻锈病致病机制, 基于RAD-seq技术检测到的SNP构建高密度遗传图谱, 检测到2个新的致病基因, 提高了对该病原体致病机制的认识[107]; 亚麻[108]和苎麻[109]也都利用GBS技术构建了高密度遗传图谱, 分别定位了株高和纤维相关QTL。
3.4 麻类作物品种改良的发展趋势
目前的麻类作物育种计划仍是依赖于将分子标记选择整合到常规育种方案中的表型选择, 即常规育种与生物技术相结合。过去, 麻类作物的育种目标大多是高产优质的纤维品种培育。但随着社会发展, 市场需求与产品多样化对麻类育种工作者提出新的挑战, 需要选育出高产高效、抗逆抗病、适宜轻简化机械化、优质专用的多用途麻类作物新品种。
一是麻类纤维用途不能只是局限于纺织业。由于麻类纤维可自然降解, 因此以麻纤维为主要原料可制作环保型的包装带或可降解地膜等。麻类纤维具有纤维强力好的特点, 其地膜的抗拉强力大于普通地膜, 适宜机械铺膜, 不仅能减少塑料薄膜导致的白色污染, 而且能减轻劳动强度和生产成本。麻纤维还能用于建筑或家饰板材等用途。近年来关于麻纤维复合材料的研究越来越多, 这是由于与玻璃纤维复合材料相比麻纤维复合材料具有密度低、隔音效果好、价格低和人体亲和性好等优点。因此培育出更适合用于制作复合材料的新品种也是当下麻类作物育种的一大趋势。
二是为了适应现代机械化, 麻类作物育种需要满足轻简栽培需要。直播和机械化等轻简栽培技术能大大降低劳动成本, 目前已经成为水稻、玉米等作物的主流生产技术, 然而麻类作物想要适应轻简栽培还需要克服易倒伏和麻秆易断等问题。为满足麻类作物适应现代机械化, 育种家们需要培育出茎秆粗壮、耐密植、抗倒伏能力强和高产优质抗病的麻类作物新品种。对于短日照喜温类型麻类作物, 宜采取“南种北植”来延长其营养生长期, 提高纤维产量。
三是为了“不与粮争好地”, 培育适合逆境农业的品种。麻类作物在贫瘠土壤的种植, 能具有更强的生产潜能, 这是棉花所不能比拟的, 培育适合逆境农业的新品种不仅仅是要求麻类作物能在不良环境下保持正常生长, 更要稳定的达到高产和优产的特点。因此, 麻类作物育种工作要更注重耐盐碱、抗旱、重金属吸附和抗病虫等性能。
四是根据不同麻类作物特异性状, 培育不同用途的专用品种。如菜用黄麻、高大麻二酚(CBD)大麻品种、籽用大麻和籽用亚麻等。福建农林大学针对福建春夏蔬菜淡季的缺口, 以中晚熟、高产、食用品质优、抗炭疽病为选育目标, 着力开展菜用黄麻新品种的选育, 最终选育出福农系列菜用黄麻, 主食嫩茎和幼叶, 口感极佳。大麻素作为大麻独有的一类次生代谢物, 具有重要的医用价值, 培育高CBD大麻品种将一直是大麻遗传改良的重点方向, 这也是近年来大麻产业蓬勃发展的重要原因之一。亚麻籽油营养丰富, 不仅含α-亚麻酸这一人体所必须的脂肪酸, 而且亚油酸等不饱和脂肪酸含量高, 对皮肤具有优良的亲和力和渗透力, 能用于护肤产品原料, 为满足市场需求, 培育专用的籽用亚麻是亚麻育种工作必不可少的。
4 展望
从优良品种到地方品种和野生近缘种的种质资源, 将仍然是任何育种计划的基础。随着麻类作物参考基因组测序完成, 如何高效挖掘这些麻类种质资源的遗传变异和重要应用价值基因位点信息并应用到遗传改良, 值得探索。NGS革命的第一个效果是降低每个测试数据的标记成本[104], 在琼脂糖凝胶或毛细管测序仪上运行的SSRs或CAPS标记要比在高通量平台上运行的SNPs昂贵得多。然后, 开发一种全基因组选择的育种方法, 如GS[96], 正在成为可能; 并将有助于设计新的植物品种, 不仅应用于少数特定的农艺性状, 而且应用于基因组中几乎所有的基因位点; 以一种成本合理和相对快速的方式, 有望给下一代育种做出显著的贡献。目前基因组选择育种只是在水稻和玉米等主要农作物中有所报道,若能将此育种方法应用于麻类作物, 必将加快麻类作物育种步伐。预计在不久的将来, NGS技术、生物信息学和表型自动化等方法在种质资源的遗传多样性中进一步应用。建立基于优异基因资源挖掘与种质创新的高通量基因型-表型数据库, 创制种质资源管理和共享平台。创新并集成分子标记辅助选择、GS、转基因等技术, 建立高效的快速育种技术体系, 将彻底改变麻类作物育种策略, 以实现更有效的遗传改良。
麻类作物作为第五大作物群, 其遗传改良越来越受益于基因组学的发展, 通过基因组学能发现一批已知位点与功能的基因资源, 从而解决育种亲本贫乏和选择效率低的问题, 同时还能供转基因育种使用, 能大幅度推动主要麻类作物遗传改良。但应该指出的是, 这些重要基因以及位点的信息绝大数是基于或依赖于模式植物的同源基因信息。麻类作物纤维相对于模式植物而言, 有其独特之处。同样地, 麻类作物响应各种非生物胁迫和氮肥等高效利用等方面也有其特异之处。以红麻耐盐碱为例, 耐盐红麻品种在400 mmol L-1的NaCl浓度下可正常生长。完成麻类作物参考基因组图谱绘制, 采用正向和反向遗传学策略挖掘麻类作物的纤维和重要性状的作用机制值得关注, 可开展麻类作物种质资源形成和演化机制研究, 系统解析纤维产量、纤维品质、抗病耐逆等农艺性状形成的分子基础。主要麻类作物特异性状的相关基因有所报道, 但功能基因鉴定仍较困难, 如工业大麻性别相关的基因, 这也与功能标记的缺乏有关。同时, 在利用这些基因进行麻类作物遗传改良的过程中还面临着遗传转化体系及其基因编辑体系构建等挑战, 如在CRISPR-Cas9基因编辑系统中, 挖掘适用于特定麻类作物的内源U6启动子并建立相应的基因编辑体系, 仍需要摸索与优化。克服这些挑战将能显著提高主要麻类作物遗传改良效率。
[1] 熊和平. 麻类作物育种学. 北京: 中国农业科学技术出版社, 2008. pp 5–8. Xiong H P. Breeding Sciences of Bast and Leaf Fiber Crops. Beijing: China Agricultural Science and Technology Press, 2008. pp 5–8 (in Chinese).
[2] 张力岚, 王俊, 万雪贝, 徐益, 张列梅, 方平平, 祁建民, 张立武. 主要麻类作物的ITS序列分析与系统进化. 作物学报, 2017, 43: 862–874. Zhang L L, Wang J, Wan X B, Xu Y, Zhang L M, Fang P P, Qi J M, Zhang L W. Analysis of internal transcribed spacers (ITS) sequences and phylogenetics of main bast fiber crops., 2017, 43: 862–874 (in Chinese with English abstract).
[3] 贾继增, 高丽锋, 赵光耀, 周文彬, 张卫建. 作物基因组学与作物科学革命. 中国农业科学, 2015, 48: 3316–3332. Jia J Z, Gao L F, Zhao G Y, Zhou W B, Zhang W J. Crop genomics and crop science revolutions., 2015, 48: 3316–3332 (in Chinese with English abstract).
[4] Initiative T A G. Analysis of the genome sequence of the flowering plant., 2000, 408: 796–815.
[5] Goff S A, Ricke D, Lan T H, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange B M, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, Sun W, Chen L, Cooper B, Park S, Wood T C, Mao L, Quail P, Wing R, Dean R, Yu Y, Zharkikh A, Shen R, Sahasrabudhe S, Thomas A, Cannings R, Gutin A, Pruss D, Reid J, Tavtigian S, Mitchell J, Eldredge G, Scholl T, Miller R M, Bhatnagar S, Adey N, Rubano T, Tusneem N, Robinson R, Feldhaus J, Macalma T, Oliphant A, Briggs S. A draft sequence of the rice genome (L. ssp.)., 2002, 296: 92–100.
[6] Islam M S, Saito J A, Emdad E M, Ahmed B, Islam M M, Halim A, Hossen Q M, Hossain M Z, Ahmed R, Hossain M S, Kabir S M, Khan M S, Khan M M, Hasan R, Aktar N, Honi U, Islam R, Rashid M M, Wan X, Hou S, Haque T, Azam M S, Moosa M M, Elias S M, Hasan A M, Mahmood N, Shafiuddin M, Shahid S, Shommu N S, Jahan S, Roy S, Chowdhury A, Akhand A I, Nisho G M, Uddin K S, Rabeya T, Hoque S M, Snigdha A R, Mortoza S, Matin S A, Islam M K, Lashkar M Z, Zaman M, Yuryev A, Uddin M K, Rahman M S, Haque M S, Alam M M, Khan H, Alam M. Comparative genomics of two jute species and insight into fibre biogenesis., 2017, 3: 16223.
[7] 张立武. 基于染色体级别参考基因组和重测序的黄麻重要性状GWAS分析. 见: 2018全国植物生物学大会论文集. 山东泰安, 2018. p 96. Zhang L W. GWAS analysis of jute important traits based on chromosome-level reference genome and resequencing. In: Proceedings of 2018 National Congress of Plant Biology. Tai’an, Shandong, China, 2018. p 96 (in Chinese).
[8] Zhang L W, Xu Y, Zhang X T, Ma X K, Zhang L L, Liao Z Y, Zhang Q, Wan X B, Cheng Y, Zhang J S, Li D X, Zhang L M, Xu J T, Tao A F, Lin L H, Fang P P, Chen S, Qi R, Xu X M, Qi J, Ming R. The genome of kenaf (L.) provides insights into bast fibre and leaf shape biogenesis., 2020, 18: 1796–1809.
[9] Liu C, Zeng L B, Zhu S Y, Wu L Q, Wang Y Z, Tang S W, Wang H W, Zheng X, Zhao J, Chen X R, Dai Q Z, Liu T M. Draft genome analysis provides insights into the fiber yield, crude protein biosynthesis, and vegetative growth of domesticated ramie (L. Gaud.)., 2018, 25: 173–181.
[10] Luan M B, Jian J B, Chen P, Chen J H, Chen J H, Gao Q, Gao G, Zhou J H, Chen K M, Guang X M, Chen J K, Zhang Q Q, Wang X F, Fang L, Sun Z M, Bai M Z, Fang X D, Zhao S C, Xiong H P, Yu C M, Zhu A G. Draft genome sequence of ramie,(L.) Gaudich., 2018, 18: 639–645.
[11] Wang Z, Hobson N, Galindo L, Zhu S, Shi D, McDill J, Yang L, Hawkins S, Neutelings G, Datla R, Lambert G, Galbraith D W, Grassa C J, Geraldes A, Cronk Q C, Cullis C, Dash P K, Kumar P A, Cloutier S, Sharpe A G, Wong G K, Wang J, Deyholos M K. The genome of flax () assembledfrom short shotgun sequence reads., 2012, 72: 461–473.
[12] Zhang J P, Qi Y N, Wang L M, Wang L L, Yan X C, Dang Z, Li W J, Zhao W, Pei X W, Li X M, Liu M, Tan M L, Wang L, Long Y, Wang J, Zhang X W, Dang Z H, Zheng H K, Liu T M. Genomic comparison and population diversity analysis provide insights into the domestication and improvement of flax., 2020, 23: 100967.
[13] Bakel H V, Stout J M, Cote A G, Tallon C M, Sharpe A G, Hughes T R, Page J E. The draft genome and transcriptome of., 2011, 12: R102.
[14] Gao S, Wang B, Xie S, Xu X, Zhang J, Pei L, Yu Y, Yang W, Zhang Y. A high-quality reference genome of wild., 2020, 7: 73.
[15] Fang S S, Zhang L M, Qi J M, Zhang L W.assembly of chloroplast genomes ofandyields species-specific InDel markers., 2021, 9: 216–226.
[16] Cheng Y, Zhang L M, Qi J M, Zhang L W. Complete chloroplast genome sequence ofand comparative analysis of the Malvaceae family., 2020, 11: 227.
[17] Zhang L W, Ming R, Zhang J S, Tao A F, Fang P P, Qi J M.transcriptome sequence and identification of major bast-related genes involved in cellulose biosynthesis in jute (L.)., 2015, 16: 1062.
[18] Yang Z M, Wu Y P, Dai Z G, Chen X J, Wang H Q, Yang S, Xie D W, Tang Q, Cheng C H, Xu Y, Deng C H, Liu C, Chen J Q, Su J G. Comprehensive transcriptome analysis and tissue-specific profiling of gene expression in jute (L.)., 2020, 146: 112101.
[19] Chakraborty A, Sarkar D, Satya P, Karmakar P G, Singh N K. Pathways associated with lignin biosynthesis in lignomaniac jute fibres., 2015, 290: 1523–1542.
[20] Samanta P, Sadhukhan S, Basu A. Identification of differentially expressed transcripts associated with bast fibre development inby suppression subtractive hybridization., 2014, 241: 371–385.
[21] Yang Z M, Yan A, Lu R K, Dai Z G, Tang Q, Cheng C H, Xu Y, Su J G.transcriptome sequencing of two cultivated jute species under salinity stress., 2017, 12: e0185863.
[22] Yang Z M, Dai Z G, Lu R K, Wu B B, Tang Q, Xu Y, Cheng C H, Su J G. Transcriptome analysis of two species of jute in response to polyethylene glycol (PEG)-induced drought stress., 2017, 7: 16565.
[23] Zhang G Y, Shan S L, Wu Y B, Huang S Q, Li D F, Deng J L, Qi J M. Thegene is involved in the formation of chloroplast stromules and other physiological processes in jute (L.)., 2019, 141: 111781.
[24] Zhang L W, Wan X B, Xu J T, Lin L H, Qi J M.assembly of kenaf () transcriptome using Illumina sequencing for gene discovery and marker identification., 2015, 35: 192.
[25] Ryu J, Kwon S J, Sung S Y, Kim W J, Kim D S, Ahn J W, Kim J B, Kim S H, Ha B K, Kang S Y. Molecular cloning, characterization, and expression analysis of lignin biosynthesis genes from kenaf (L.)., 2015, 38: 59–67.
[26] 黄枝妙. 红麻成花光周期调控相关基因和的克隆与表达. 福建农林大学硕士学位论文, 福建福州, 2014. Huang Z M. Cloning and Expression Analysis of Photoperiodic Flowering Related Genesandin Kenaf. MS Thesis of Fujian Agriculture and Forestry University, Fuzhou, Fujian, China, 2014 (in Chinese with English abstract).
[27] Li H, Li D F, Chen A G, Tang H J, Li J J, Huang S Q. RNA-seq for comparative transcript profiling of kenaf under salinity stress., 2016, 130: 365–372.
[28] 李辉, 李德芳, 陈安国, 唐慧娟, 李建军, 黄思齐. 盐和干旱胁迫下红麻基因的克隆及表达特征. 农业生物技术学报, 2017, 25: 1970–1978. Li H, Li D F, Chen A G, Tang H J, Li J J, Huang S Q. Cloning and expression characteristics ofgene under salt and drought stress in kenaf ()., 2017, 25: 1970–1978 (in Chinese with English abstract).
[29] 潘根, 赵立宁, 陈安国, 李建军, 黄思齐, 唐慧娟, 常丽, 邓勇, 李德芳. 红麻基因的克隆与表达特征分析. 中国麻业科学, 2018, 40(4): 145–150. Pan G, Zhao L N, Chen A G, Li J J, Huang S Q, Tang H J, Chang L, Deng Y, Li D F. Cloning and expression analysis of atranscript factorin kenaf., 2018, 40(4): 145–150 (in Chinese with English abstract).
[30] Wei F, Tang D, Li Z, Kashif M H, Khan A, Lu H, Jia R, Chen P. Molecular cloning and subcellular localization of sixs and their roles in response to salt and drought stress in kenaf (L.)., 2019, 52: 20.
[31] 周瑞阳, 张新, 张加强, 甘正华, 韦汉西. 红麻细胞质雄性不育系的选育及杂种优势利用取得突破. 中国农业科学, 2008, 41: 314. Zhou R Y, Zhang X, Zhang J Q, Gan Z H, Wei H X. A breakthrough in kenaf cytoplasmic male sterile lines breeding and heterosis utilization., 2008, 41: 314 (in Chinese with English abstract).
[32] Zhao Y H, Chen P, Liao X F, Zhou B J, Liao J, Huang Z P, Kong X J, Zhou R Y. A comparative study of thegene between a cytoplasmic male sterile line and its maintainer line and further development of a molecular marker specific for male sterile cytoplasm in kenaf (L.)., 2013, 32: 969–976.
[33] Zhao Y H, Liao X F, Huang Z P, Chen P, Zhou B J, Liu D M, Kong X J, Zhou R Y. Expression of kenaf mitochondrial chimeric genescauses male sterility in transgenic tobacco plants., 2014, 26: 495–500.
[34] 赵艳红, 廖小芳, 赵洪涛, 黄其椿, 唐兴富, 李初英, 周瑞阳. 红麻线粒体基因克隆及不育细胞质分子标签的利用. 南方农业学报, 2015, 46: 964–970. Zhao Y H, Liao X F, Zhao H T, Huang Q C, Tang X F, Li C Y, Zhou R Y. Cloning mitochondrial geneand utilization molecular marker associated with male sterile cytoplasm in kenaf., 2015, 46: 964–970 (in Chinese with English abstract).
[35] Chen P, Ran S M, Li R, Huang Z P, Qian J H, Yu M L, Zhou R Y. Transcriptomeassembly and differentially expressed genes related to cytoplasmic male sterility in kenaf (L.)., 2014, 34: 1879–1891.
[36] Zhao Y H, Liao X F, Zhou B J, Zhao H T, Zhou Y Y, Zhou R Y. Mutation in the coding sequence ofare associated with male sterile cytoplasm in kenaf (L.)., 2015, 207: 169–175.
[37] 彭双双. 红麻UG93A雄性不育相关基因互作蛋白的鉴定与验证. 广西大学硕士学位论文, 广西南宁, 2019. Peng S S. Identification and Validation ofInteracton Protein of UG93A Male Eterility Related Gene in Kenaf. MS Thesis of Guangxi University, Nanning, Guangxi, China, 2019 (in Chinese with English abstract).
[38] 吴丹, 唐冬英, 李新梅, 李丽, 赵小英, 刘选明. F-box蛋白在植物生长发育中的功能研究进展. 生命科学研究, 2015, 19: 362–367. Wu D, Tang D Y, Li X M, Li L, Zhao X Y, Liu X M. Progresses on F-box protein function in plant growth and development., 2015, 19: 362–367 (in Chinese with English abstract).
[39] 陈励虹, 周步进, 周瑞阳. 红麻基因克隆及其表达载体构建. 南方农业学报, 2017, 48: 1343–1350. Chen L H, Zhou B J, Zhou R Y. Cloning ofgene inL. and construction of its expression vector., 2017, 48: 1343–1350 (in Chinese with English abstract).
[40] Tang D, Wei F, Kashif M H, Munsif F, Zhou R. Identification and analysis of RNA editing sites in chloroplast transcripts of kenaf (L.)., 2019, 9: 361.
[41] Liu T M, Zhu S Y, Tang Q M, Chen P, Yu Y T, Tang S W.assembly and characterization of transcriptome using Illumina paired-end sequencing and identification ofgene in ramie (L. Gaud)., 2013, 14: 125.
[42] 蒋杰, 揭雨成, 周清明, 周精华, 朱守晶, 邢虎成, 钟英丽. 苎麻纤维素合酶基因全长cDNA的克隆与表达分析. 植物遗传资源学报, 2012, 13: 851–857. Jiang J, Jie Y C, Zhou Q M, Zhou J H, Zhu S J, Xing H C, Zhong Y L. Full-length cDNA cloning and express analysis ofin ramie., 2012, 13: 851–857 (in Chinese with English abstract).
[43] 刘昱翔, 陈建荣, 彭彦, 黄妤, 赵燕, 黄丽华, 郭清泉, 张学文. 两种苎麻纤维素合酶基因cDNA序列的克隆及表达. 作物学报, 2014, 40: 1925–1935. Liu Y X, Chen J R, Peng Y, Huang Y, Zhao Y, Huang L H, Guo Q Q, Zhang X W. cDNA cloning and expression of two cellulose synthase genes from., 2014, 40: 1925–1935 (in Chinese with English abstract).
[44] 刘昱翔, 陈建荣, 彭彦, 黄妤, 赵燕, 黄丽华, 郭清泉, 张学文. 苎麻纤维素合成酶基因cDNA序列的克隆与表达分析. 作物研究, 2014, 28: 472–478. Liu Y X, Chen J R, Peng Y, Huang Y, Zhao Y, Huang L H, Guo Q Q, Zhang X W. The cDNA cloning and expression analysis on cellulose synthasein., 2014, 28: 472–478 (in Chinese with English abstract).
[45] Liu T M, Tang S W, Zhu S Y, Tang Q M, Zheng X. Transcriptome comparison reveals the patterns of selection in domesticated and wild ramie (L. Gaud.)., 2014, 86: 85–92.
[46] Chen J R, Rao J, Wang Y Z, Zeng Z, Liu F, Tang Y H, Chen X R, Liu C, Liu T M. Integration of quantitative trait loci mapping and expression profiling analysis to identify genes potentially involved in ramie fiber lignin biosynthesis., 2019, 10: 842.
[47] Tang Y H, Liu F, Xing H C, Mao K Q, Chen G, Guo Q Q, Chen J R. Correlation Analysis of lignin accumulation and expression of key genes involved in lignin biosynthesis of ramie ()., 2019, 10: 389.
[48] 李富. 苎麻、和基因的克隆及功能研究. 吉首大学硕士学位论文, 湖南湘西, 2019. Li F. Cloning and Functional Characterization of,Genes in ramie. MS Thesis of Jishou University, Xiangxi, Hunan, China, 2019 (in Chinese with English abstract).
[49] Xie J, Li J, Jie Y, Xie D, Yang D, Shi H, Zhong Y. Comparative transcriptomics of stem bark reveals genes associated with bast fiber development inL. Gaud. (ramie)., 2020, 21: 40.
[50] 付莉莉, 刘头明, 朱四元, 汤清明, 唐守伟. 苎麻转录因子的序列分析. 中国麻业科学, 2013, 35(3): 113–117. Fu L L, Liu T M, Zhu S Y, Tang Q M, Tang S W. Sequence analysis oftranscription factor in ramie., 2013, 35(3): 113–117 (in Chinese with English abstract).
[51] 薛丽君, 周精华, 邢虎成. 苎麻氧化酶基因()的克隆及表达. 中国农业科学, 2013, 46: 2377–2385. Xue L J, Zhou J H, Xing H C. Cloning and characterization ofoxidase gene () from ramie ()., 2013, 46: 2377–2385 (in Chinese with English abstract).
[52] 周精华, 揭雨成, 邢虎成, 钟英丽, 余伟林. 苎麻转录因子基因的克隆与表达特征分析. 中国农业科学, 2013, 46: 1314–1322. Zhou J H, Jie Y C, Xing H C, Zhong Y L, Yu W L. Cloning and characterization of thetranscription factor gene from ramie (L.)., 2013, 46: 1314–1322 (in Chinese with English abstract).
[53] 余伟林, 钟英丽, 揭雨成, 周清明, 周精华, 朱守晶. 苎麻基因的克隆与表达. 农业生物技术学报, 2014, 22: 27–36. Yu W L, Zhong Y L, Jie Y C, Zhou Q M, Zhou J H, Zhu S J. Molecular cloning and characterization ofgene from ramie ()., 2014, 22: 27–36 (in Chinese with English abstract).
[54] 郑建树, 喻春明, 陈平, 王延周, 谭龙涛, 陈继康, 朱涛涛, 卢凌霄, 朱娟娟, 段叶辉, 熊和平. 苎麻谷氨酰胺合成酶等位基因的克隆及其转基因烟草特性. 中国农业科学, 2014, 47: 3348–3358. Zheng J S, Yu C M, Chen P, Wang Y Z, Tan L T, Chen J K, Zhu T T, Lu L X, Zhu J J, Duan Y H, Xiong H P. Cloning of glutamine synthetaseallele genes from ramie (L.) and study on gene-transforming tobacco., 2014, 47: 3348–3358 (in Chinese with English abstract).
[55] 朱守晶, 石朝艳, 余伟林, 周精华, 揭雨成. 苎麻植物螯合肽合成酶基因的克隆和表达特性分析. 植物遗传资源学报, 2014, 15: 582–588. Zhu S J, Shi C Y, Yu W L, Zhou J H, Jie Y C. Cloning and characterization of thegene from ramie (L.)., 2014, 15: 582–588 (in Chinese with English abstract).
[56] 朱守晶, 史文娟. 苎麻转录因子基因的克隆及表达分析. 西北植物学报, 2019, 39: 422–429. Zhu S J, Shi W J. Cloning and expression pattern analysis oftranscription factor gene in ramie., 2019, 39: 422–429 (in Chinese with English abstract).
[57] 朱守晶, 史文娟. 苎麻镉响应基因的克隆和表达分析. 江苏农业学报, 2019, 35: 262–270. Zhu S J, Shi W J. Cloning and expression analysis of cadmium-responsive genefrom ramie (L.)., 2019, 35: 262–270 (in Chinese with English abstract).
[58] 尹伟丹, 马玉申, 汪娅梅, 揭雨成, 邢虎成. 苎麻基因的克隆与表达特异性分析. 分子植物育种, 2019, 18: 6298–6304. Yin W D, Ma Y S, Wang Y M, Jie Y C, Xing H C. Cloning and expression specificity analysis ofgene in ramie., 2019, 18: 6298–6304 (in Chinese with English abstract).
[59] Chen K, Yu Y, Sun K, Xiong H, Yu C, Chen P, Chen J, Gao G, Zhu A. The miRNAome of ramie (L.): identification, expression, and potential roles of novel microRNAs in regulation of cadmium stress response., 2018, 18: 369.
[60] 段继强, 李建永, 杜光辉, 梁雪妮, 刘飞虎. 苎麻线粒体基因和与细胞质雄性不育相关性分析. 中国农业科学, 2009, 42: 434–445. Duan J Q, Li J Y, Du G H, Liang X N, Liu F H. Relationship of mitochondrial genesandwith cytoplasmic male sterility in ramie., 2009, 42: 434–445 (in Chinese with English abstract).
[61] Liu L X, Zhang S W, Duan J Q, Du G H, Liu F H. Mitochondrial genesandcloned and characterized from ramie ((L.) Gaud.) and their relationship with cytoplasmic male sterility., 2011, 30: 23–32.
[62] Pydiura N A, Bayer G Y, Galinousky D V, Yemets A I, Pirko Y V, Padvitski T A, Anisimova N V, Khotyleva L V, Kilchevsky A V, Blume Y B. Bioinformatic search for cellulose synthase genes in flax () and their phylogenetic analysis., 2015, 49: 279–287.
[63] Chantreau M, Chabbert B, Billiard S, Hawkins S, Neutelings G. Functional analyses of cellulose synthase genes in flax () by virus-induced gene silencing., 2015, 13: 1312–1324.
[64] 袁红梅, 郭文栋, 赵丽娟, 于莹, 吴建忠, 张利国, 程莉莉, 赵东升, 吴广文, 关凤芝. 亚麻纤维素合酶超基因家族的生物信息学及表达分析. 中国农业科学, 2016, 49: 4656–4668. Yuan H M, Guo W D, Zhao L J, Yu Y, Wu J Z, Zhang L G, Cheng L L, Zhao D S, Wu G W, Guan F Z. Bioinformatics and expression analysis of the cellulose synthase supergene family in flax., 2016, 49: 4656–4668 (in Chinese with English abstract).
[65] Galinousky D, Padvitski T, Bayer G, Pirko Y, Pydiura N, Anisimova N, Nikitinskaya T, Khotyleva L, Yemets A, Kilchevsky A, Blume Y. Expression analysis of cellulose synthase and main cytoskeletal protein genes in flax (L.)., 2017, 43: 1065–1071.
[66] Gorshkov O, Mokshina N, Gorshkov V, Chemikosova S, Gogolev Y, Gorshkova T. Transcriptome portrait of cellulose-enriched flax fibres at advanced stage of specialization., 2016, 93: 431–449.
[67] 黄文功, 康庆华, 姜卫东, 姚玉波, 詹亚光. 木质素合成酶基因反义对亚麻的转化及检测. 中国麻业科学, 2016, 38(2): 54–57. Huang W G, Kang Q H, Jiang W D, Yao Y B, Zhan Y G. Transformation and detection ofwithgene., 2016, 38(2): 54–57 (in Chinese with English abstract).
[68] Wróbel-Kwiatkowska M, Kropiwnicki M, Żebrowski J, Beopoulos A, Dymińska L, Hanuza J, Rymowicz W. Effect of mcl-PHA synthesis in flax on plant mechanical properties and cell wall composition., 2018, 28: 77–90.
[69] Markulin L, Corbin C, Renouard S, Drouet S, Durpoix C, Mathieu C, Lopez T, Auguin D, Hano C, Laine E. Characterization of, a flax transcription factor promoting secoisolariciresinol biosynthesis in response to Fusarium oxysporum elicitors inL. hairy roots., 2019, 250: 347–366.
[70] 谢冬微, 路颖, 赵德宝, 杨学, 粟建光, 孙健. 亚麻类抗病基因家族全基因组分析. 中国麻业科学, 2015, 37(3): 113–119. Xie D W, Lu Y, Zhao D B,Yang X, Su J G, Sun J. Genome-wide analysis ofresistance genes in flax., 2015, 37(3): 113–119 (in Chinese with English abstract).
[71] Saha D, Mukherjee P, Dutta S, Meena K, Sarkar S K, Mandal A B, Dasgupta T, Mitra J. Genomic insights intos as candidate genes for high-temperature stress adaptation and gene editing with minimal off-target effects in flax., 2019, 9: 5581.
[72] Dmitriev A A, Kudryavtseva A V, Krasnov G S, Koroban N V, Speranskaya A S, Krinitsina A A, Belenikin M S, Snezhkina A V, Sadritdinova A F, Kishlyan N V, Rozhmina T A, Yurkevich O Y, Muravenko O V, Bolsheva N L, Melnikova N V. Gene expression profiling of flax (L.) under edaphic stress., 2016, 16: 237.
[73] Zyablitsin A V, Dmitriev A A, Krasnov G S, Bolsheva N L, Rozhmina T A, Muravenko O V, Fedorova M S, Snezhkina A V, Kudryavtseva A V, Melnikova N V.gene is involved in flax response to high soil acidity and aluminum exposure., 2018, 52: 514–519.
[74] Krasnov G S, Dmitriev A A, Zyablitsin A V, Rozhmina T A, Zhuchenko A A, Kezimana P, Snezhkina A V, Fedorova M S, Novakovskiy R O, Pushkova E N, Povkhova L V, Bolsheva N L, Kudryavtseva A V, Melnikova N V. Aluminum responsive genes in flax (L.)., 2019, 2019: 1–11.
[75] Soto-Cerda B J, Cloutier S, Gajardo H A, Aravena G, Quian R. Identifying drought-resilient flax genotypes and related-candidate genes based on stress indices, root traits and selective sweep., 2019, 215: 1–16.
[76] Wu W, Nemri A, Blackman L M, Catanzariti A M, Sperschneider J, Lawrence G J, Dodds P N, Jones D A, Hardham A R. Flax rust infection transcriptomics reveals a transcriptional profile that may be indicative for rustgenes., 2019, 14: e0226106.
[77] 李晓薇, 戢舒涵, 赵旭, 张百兵, 王法微, 李海燕. 亚麻芥未成熟胚中磷脂酶基因家族的转录组学分析. 中国油料作物学报, 2018, 40: 793–800. Li X W, Ji S H, Zhao X, Zhang B B, Wang F W, Li H Y. Transcriptome analysis of phospholipase gene family in immature embryo of., 2018, 40: 793–800 (in Chinese with English abstract).
[78] 李闻娟, 齐燕妮, 王利民, 党照, 赵利, 赵玮, 谢亚萍, 王斌, 张建平, 李淑洁. 不同胡麻品种TAG合成途径关键基因表达与含油量、脂肪酸组分的相关性分析. 草业学报, 2019, 28(1): 138–149. Li W J, Qi Y N, Wang L M, Dang Z, Zhao L, Zhao W, Xie Y P, Wang B, Zhang J P, Li S J. Correlation between oil content or fatty acid composition and expression levels of genes involved in TAG biosynthesis in flax., 2019, 28(1): 138–149 (in Chinese with English abstract).
[79] Xie D, Dai Z, Yang Z, Tang Q, Deng C, Xu Y, Wang J, Chen J, Zhao D, Zhang S, Zhang S, Su J. Combined genome-wide association analysis and transcriptome sequencing to identify candidate genes for flax seed fatty acid metabolism., 2019, 286: 98–107.
[80] Behr M, Legay S, Žižková E, Motyka V, Dobrev P I, Hausman J F, Lutts S, Guerriero G. Studying secondary growth and bast fiber development: the hemp hypocotyl peeks behind the wall., 2016, 7: 1733.
[81] Zhong R, Ye Z H. Secondary cell walls: biosynthesis, patterned deposition and transcriptional regulation., 2015, 56: 195–214.
[82] Zhong R, Lee C, Zhou J, McCarthy R L, Ye Z H. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in., 2008, 20: 2763–2782.
[83] Han L B, Li Y B, Wang H Y, Wu X M, Li C L, Luo M, Wu S J, Kong Z S, Pei Y, Jiao G L, Xia G X. The dual functions ofin cell elongation and secondary wall formation in developing cotton fibers., 2013, 25: 4421–4438.
[84] MacMillan C P, Taylor L, Bi Y, Southerton S G, Evans R, Spokevicius A. The fasciclin-like arabinogalactan protein family of Eucalyptus grandis contains members that impact wood biology and biomechanics., 2015, 206: 1314–1327.
[85] Alexander S P H. Therapeutic potential of cannabis-related drugs., 2016, 64: 157–166.
[86] 陈璇, 张庆滢, 郭蓉, 郭孟璧, 许艳萍, 杨明, 郭鸿彦. 不同发育时期大麻素合成相关酶基因表达特征与大麻素含量的相关分析. 分子植物育种, 2018, 16: 583–590. Chen X, Zhang Q Y, Guo R, Guo M B, Xu Y P, Yang M, Guo H Y. Correlation analysis between gene expression characteristics of related enzymes in cannabinoids biosynthesis and cannabinoids content at different developmental stages ofL., 2018, 16: 583–590 (in Chinese with English abstract).
[87] Zager J J, Lange I, Srividya N, Smith A, Lange B M. Gene networks underlying cannabinoid and terpenoid accumulation in., 2019, 180: 1877–1897.
[88] Duraisamy G S, Mishra A K, Kocábek T, Matoušek J. Activation of polyketide synthase gene promoter inby heterologous transcription factors derived from., 2018, 62: 250–260.
[89] Laverty K U, Stout J M, Sullivan M J, Shah H, Gill N, Holbrook L, Deikus G, Sebra R, Hughes T R, Page J E, Bakel H V. A physical and genetic map ofidentifies extensive rearrangements at the THC/CBD acid synthase loci., 2019, 29: 146–156.
[90] Prentout D, Razumova O, Rhoné B, Badouin H, Henri H, Feng C, Käfer J, Karlov G, Marais G A B. An efficient RNA-seq-based segregation analysis identifies the sex chromosomes of., 2020, 30: 164–172.
[91] 陆朝福, 朱立煌. 植物育种中的分子标记辅助选择. 生物工程进展, 1995, 15(4): 11–17. Lu C F, Zhu L H. Molecular marker assisted selection in plant breeding., 1995, 15(4): 11–17 (in Chinese).
[92] 王斌, 赵利, 赵玮. 8个地方野生亚麻资源发掘及遗传多样性分析. 分子植物育种, 2018, 17: 3755–3760. Wang B, Zhao L, Zhao W. Exploration and genetic diversity analysis of 8 local wild flax germplasm resources., 2018, 17: 3755–3760 (in Chinese with English abstract).
[93] Soorni A, Fatahi R, Haak D C, Salami S A, Bombarely A. Assessment of genetic diversity and population structure in iraniangermplasm., 2017, 7: 15668.
[94] Biswas C, Dey P, Karmakar P G, Satpathy S. Discovery of large-scale SNP markers and construction of linkage map in a RIL population of jute ()., 2015, 35: 119.
[95] Yang Z M, Yang Y X, Dai Z G, Xie D W, Tang Q, Cheng C H, Xu Y, Liu C, Deng C H, Chen J Q, Su J G. Construction of a high-resolution genetic map and identification of quantitative trait loci for salt tolerance in jute (spp.)., 2019, 19. DOI: 10.1186/s12870-019-2004-7.
[96] Meuwissen T H E, Hayes B J, Goddard M E. Prediction of total genetic value using genome-wide dense marker maps., 2001, 157: 1819–1829.
[97] Morrell P L, Buckler E S, Ross-Ibarra J. Crop genomics: advances and applications., 2011, 13: 85–96.
[98] Riedelsheimer C, Czedik-Eysenberg A, Grieder C, Lisec J, Technow F, Sulpice R, Altmann T, Stitt M, Willmitzer L, Melchinger A E. Genomic and metabolic prediction of complex heterotic traits in hybrid maize., 2012, 44: 217–220.
[99] Xu S Z, Xu Y, Gong L, Zhang Q F. Metabolomic prediction of yield in hybrid rice., 2016, 88: 219–227.
[100] 刘策, 孟焕文, 程智慧. 植物全基因组选择育种技术原理与研究进展. 分子植物育种, 2020, 18: 5335–5342. Liu C, Meng H W, Cheng Z H. Plant genomic selection breeding technical principle and research progress., 2020, 18: 5335–5342 (in Chinese with English abstract).
[101] 孙怡迪, 左二伟, 杨辉. 基因编辑技术的风险和机遇. 科学, 2019, 71(6): 25–28. Sun Y D, Zuo E W, Yang H. Risks and opportunities of gene editing technology., 2019, 71(6): 25–28 (in Chinese).
[102] 张喻, 江海霞, 闫文亮, 郭栋良, 杨亮杰, 叶佳丽, 王玥, 谢丽琼. CRISPR_Cas9系统敲除亚麻基因表达载体的构建. 分子植物育种, 2019, 17: 2185–2192. Zhang Y, Jiang H X, Yan W L, Guo D L, Yang L J, Ye J L, Wang Y, Xie L Q. Construction of expression vector for knocking outgene in flax by CRISPR/Cas9 system., 2019, 17: 2185–2192 (in Chinese with English abstract).
[103] 任羽, 尹俊梅, 潘红兵, 徐世松, 黄少华. 园艺植物遗传图谱的研究进展. 中国农学通报, 2012, 28(7): 89–94. Ren Y, Yin J M, Pan H B, Xu S S, Huang S H. Research progress in genetic linkage map of horticulture plant., 2012, 28(7): 89–94 (in Chinese with English abstract).
[104] 解增言, 林俊华, 谭军, 舒坤贤. DNA测序技术的发展历史与最新进展. 生物技术通报, 2010, (8): 64–70. Xie Z Y, Lin J H, Tan H, Shu K X. The history and advances of DNA sequencing technojogy., 2010, (8): 64–70 (in Chinese with English abstract).
[105] 王洋坤, 胡艳, 张天真. RAD-seq技术在基因组研究中的现状及展望. 遗传, 2014, 36: 41–49. Wang Y K, Hu Y, Zhang T Z. Current status and perspective of RAD-seq in genomic research., 2014, 36: 41–49 (in Chinese with English abstract).
[106] 张羽, 胡仁发, 周婉莹, 孙旺. 基于限制性内切酶简化基因组测序的两种主要技术. 分子植物育种, 2020, 18: 3562–3570. Zhang Y, Hu R F, Zhou W Y, Sun W. The two major technologies of sequencing based on simplified genome by restriction enzyme digestion., 2020, 18: 3562–3570 (in Chinese with English abstract).
[107] Anderson C, Khan M A, Catanzariti A M, Jack C A, Nemri A, Lawrence G J, Upadhyaya N M, Hardham A R, Ellis J G, Dodds P N, Jones D A. Genome analysis and avirulence gene cloning using a high-density RADseq linkage map of the flax rust fungus,., 2016, 17: 667.
[108] Zhang J, Long Y, Wang L, Dang Z, Zhang T, Song X, Dang Z, Pei X. Consensus genetic linkage map construction and QTL mapping for plant height-related traits in linseed flax (L.)., 2018, 18: 160.
[109] Liu C, Zhu S, Tang S, Wang H, Zheng X, Chen X, Dai Q, Liu T. QTL analysis of four main stem bark traits using a GBS-SNP-based high-density genetic map in ramie., 2017, 7: 13458.
[110] 谈静, 郭俊杰, 曾杰. 多倍体植物复杂性状全基因组关联分析研究进展. 分子植物育种, 2020, 18: 1282–1289. Tan J, Guo J J, Zeng J. Advance in genome-wide association analysis of complex traits for polyploid plants., 2020, 18: 1282–1289 (in Chinese with English abstract).
[111] Chen K, Luan M, Xiong H, Chen P, Chen J, Gao G, Huang K, Zhu A, Yu C. Genome-wide association study discovered favorable single nucleotide polymorphisms and candidate genes associated with ramet number in ramie (L.)., 2018, 18: 345.
[112] Xie D W, Dai Z G, Yang Z M, Sun J, Zhao D B, Yang X, Zhang L G, Tang Q, Su J G. Genome-wide association study identifying candidate genes influencing important agronomic traits of flax (L.) using SLAF-seq., 2018, 8: 2232.
[113] 伊六喜, 斯钦巴特尔, 冯小慧, 贾霄云, 高凤云, 周宇, 张辉. 胡麻木酚素含量的全基因组关联分析. 分子植物育种, 2020, 18: 765–771. Yi L X, Bateer S, Feng X H, Jia X Y, Gao F Y, Zhou Y, Zhang H. Genome-wide association analysis of lignan content in flax., 2020, 18: 765–771 (in Chinese with English abstract).
[114] He L, Xiao J, Rashid K Y, Yao Z, Li P, Jia G, Wang X, Cloutier S, You F M. Genome-wide association studies for pasmo resistance in flax (L.)., 2018, 9: 1982.
[115] Soto-Cerda B J, Cloutier S, Quian R, Gajardo H A, Olivos M, You F M. Genome-wide association analysis of mucilage and hull content in flax (L.) seeds., 2018, 19: 2870.
附图1 基于叶绿体基因组序列的圆果种黄麻和长果种黄麻与锦葵科植物的系统发育关系
Fig. S1 Phylogenetic relationship ofandwith species of the Malvaceae family based on the chloroplast genome sequences
附表1 主要麻类作物纤维发育候选基因信息
Table S1 Information of candidate genes for fiber development in main bast fiber crops
(续附表1)
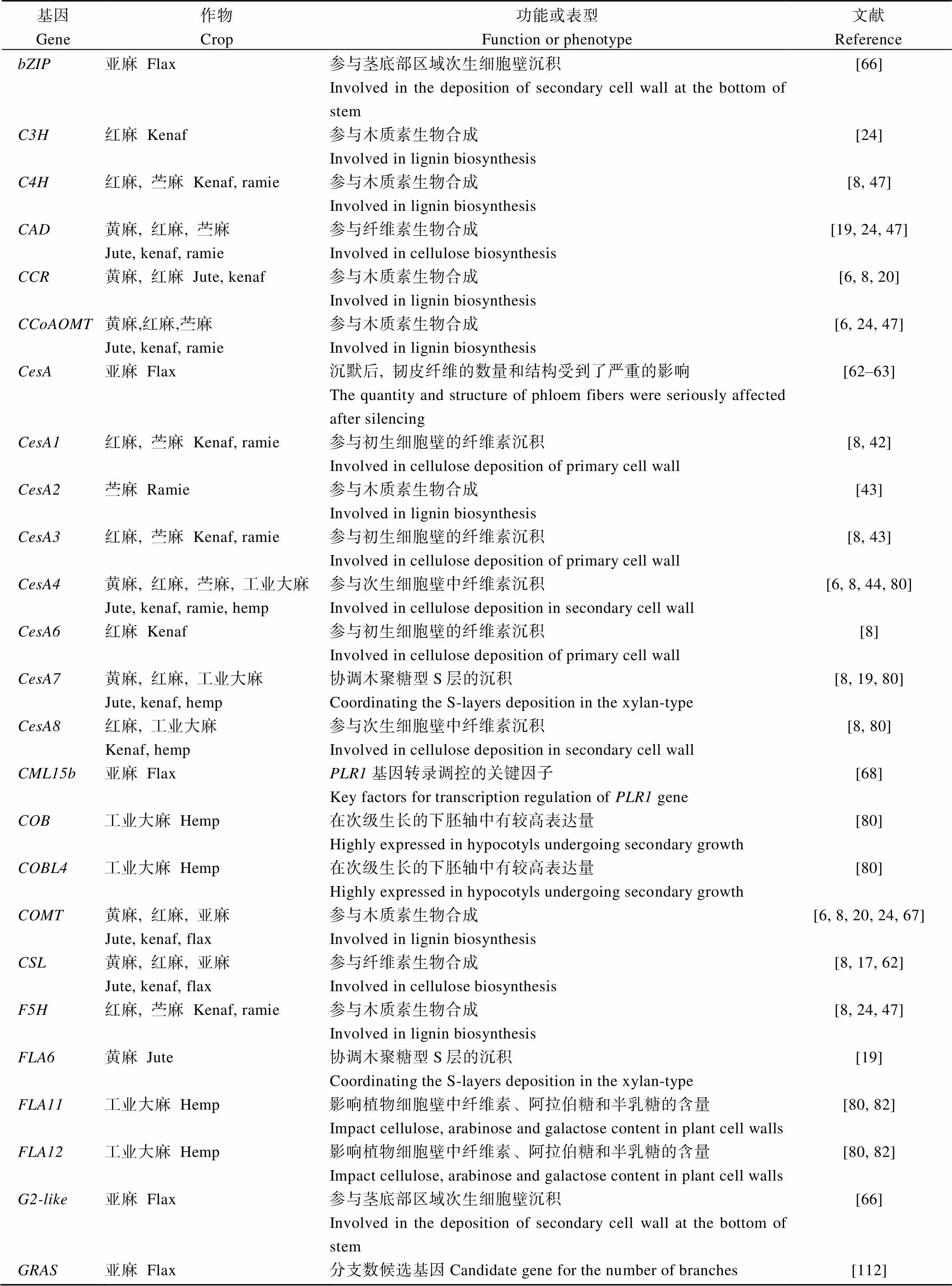
基因Gene作物Crop功能或表型Function or phenotype文献Reference bZIP亚麻 Flax参与茎底部区域次生细胞壁沉积Involved in the deposition of secondary cell wall at the bottom of stem[66] C3H红麻 Kenaf参与木质素生物合成Involved in lignin biosynthesis[24] C4H红麻, 苎麻 Kenaf, ramie参与木质素生物合成Involved in lignin biosynthesis[8, 47] CAD黄麻, 红麻, 苎麻 Jute, kenaf, ramie参与纤维素生物合成Involved in cellulose biosynthesis[19, 24, 47] CCR黄麻, 红麻 Jute, kenaf参与木质素生物合成Involved in lignin biosynthesis[6, 8, 20] CCoAOMT黄麻,红麻,苎麻 Jute, kenaf, ramie参与木质素生物合成Involved in lignin biosynthesis[6, 24, 47] CesA亚麻 Flax沉默后, 韧皮纤维的数量和结构受到了严重的影响 The quantity and structure of phloem fibers were seriously affected after silencing[62–63] CesA1红麻, 苎麻 Kenaf, ramie参与初生细胞壁的纤维素沉积 Involved in cellulose deposition of primary cell wall[8, 42] CesA2苎麻 Ramie参与木质素生物合成Involved in lignin biosynthesis[43] CesA3红麻, 苎麻 Kenaf, ramie参与初生细胞壁的纤维素沉积 Involved in cellulose deposition of primary cell wall[8, 43] CesA4黄麻, 红麻, 苎麻, 工业大麻 Jute, kenaf, ramie, hemp参与次生细胞壁中纤维素沉积 Involved in cellulose deposition in secondary cell wall[6, 8, 44, 80] CesA6红麻 Kenaf参与初生细胞壁的纤维素沉积 Involved in cellulose deposition of primary cell wall[8] CesA7黄麻, 红麻, 工业大麻 Jute, kenaf, hemp协调木聚糖型S层的沉积 Coordinating the S-layers deposition in the xylan-type[8, 19, 80] CesA8红麻, 工业大麻 Kenaf, hemp参与次生细胞壁中纤维素沉积 Involved in cellulose deposition in secondary cell wall[8, 80] CML15b亚麻 FlaxPLR1基因转录调控的关键因子 Key factors for transcription regulation of PLR1 gene[68] COB工业大麻 Hemp在次级生长的下胚轴中有较高表达量 Highly expressed in hypocotyls undergoing secondary growth[80] COBL4工业大麻 Hemp在次级生长的下胚轴中有较高表达量 Highly expressed in hypocotyls undergoing secondary growth[80] COMT黄麻, 红麻, 亚麻 Jute, kenaf, flax参与木质素生物合成Involved in lignin biosynthesis[6, 8, 20, 24, 67] CSL黄麻, 红麻, 亚麻 Jute, kenaf, flax参与纤维素生物合成Involved in cellulose biosynthesis[8, 17, 62] F5H红麻, 苎麻 Kenaf, ramie参与木质素生物合成Involved in lignin biosynthesis[8, 24, 47] FLA6黄麻 Jute协调木聚糖型S层的沉积 Coordinating the S-layers deposition in the xylan-type[19] FLA11工业大麻 Hemp影响植物细胞壁中纤维素、阿拉伯糖和半乳糖的含量 Impact cellulose, arabinose and galactose content in plant cell walls[80, 82] FLA12工业大麻 Hemp影响植物细胞壁中纤维素、阿拉伯糖和半乳糖的含量 Impact cellulose, arabinose and galactose content in plant cell walls[80, 82] G2-like亚麻 Flax参与茎底部区域次生细胞壁沉积 Involved in the deposition of secondary cell wall at the bottom of stem[66] GRAS亚麻 Flax分支数候选基因Candidate gene for the number of branches[112]
(续附表1)
(续附表1)

基因Gene作物Crop功能或表型Function or phenotype文献Reference TDIF红麻 Kenaf参与纤维形成 Involved in fiber formation[8] TOUCH4工业大麻 Hemp将木葡聚糖靶向于次生细胞壁S1层的初生-次生细胞壁交界Participate in the targeting of xyloglucan to the primary- secondary cell wall junction in the secondary cell wall S1 layer[80] UGPase黄麻 Jute参与纤维素生物合成 Involved in cellulose biosynthesis[17] UGT亚麻 Flax株高候选基因 Candidate gene for plant height[112] WAT1苎麻 Ramie高纤维品种驯化中的受选择基因Selection of highly domesticated fiber varieties[45] WLIM1工业大麻 Hemp捆绑肌动蛋白丝来促进纤维的延伸, 与PAL-box结合来促进木质素生物合成基因的木质化 Fibre extension by bundling the actin filament but also fibre lignification by promoting the lignin/lignin-like biosynthetic genes via binding to the PAL-box[80, 83] WOX4黄麻, 红麻 Jute, kenaf参与纤维素生物合成Involved in cellulose biosynthesis[6, 8] WRKY黄麻 Jute纤维改良 Fiber improvement[20] WRKY36亚麻 Flax通过与PLR1启动子中的W-box结合, 参与木质素生物合成Involved in lignin biosynthesis by binding with W-box in PLR1 promoter[69] XTH亚麻 Flax分支数候选基因Candidate gene for the number of branches[112] XTH5工业大麻 Hemp韧皮纤维的伸长依赖于XTH的活性Bast fibre extension depends on the activities of XTH[80] XTH8工业大麻 Hemp韧皮纤维的伸长依赖于XTH的活性 Bast fibre extension depends on the activities of XTH[80] XTH15工业大麻 Hemp在二次生长的较老大麻下胚轴中表达较多 More expressed in the elongating hemp hypocotyl[80] XTH22工业大麻 Hemp在二次生长的较老大麻下胚轴中表达较多 More expressed in the elongating hemp hypocotyl[80]
附表2 主要麻类作物响应非生物胁迫候选基因信息
Table S2 Information of candidate genes for abiotic stress response in main bast fiber crops
(续附表2)
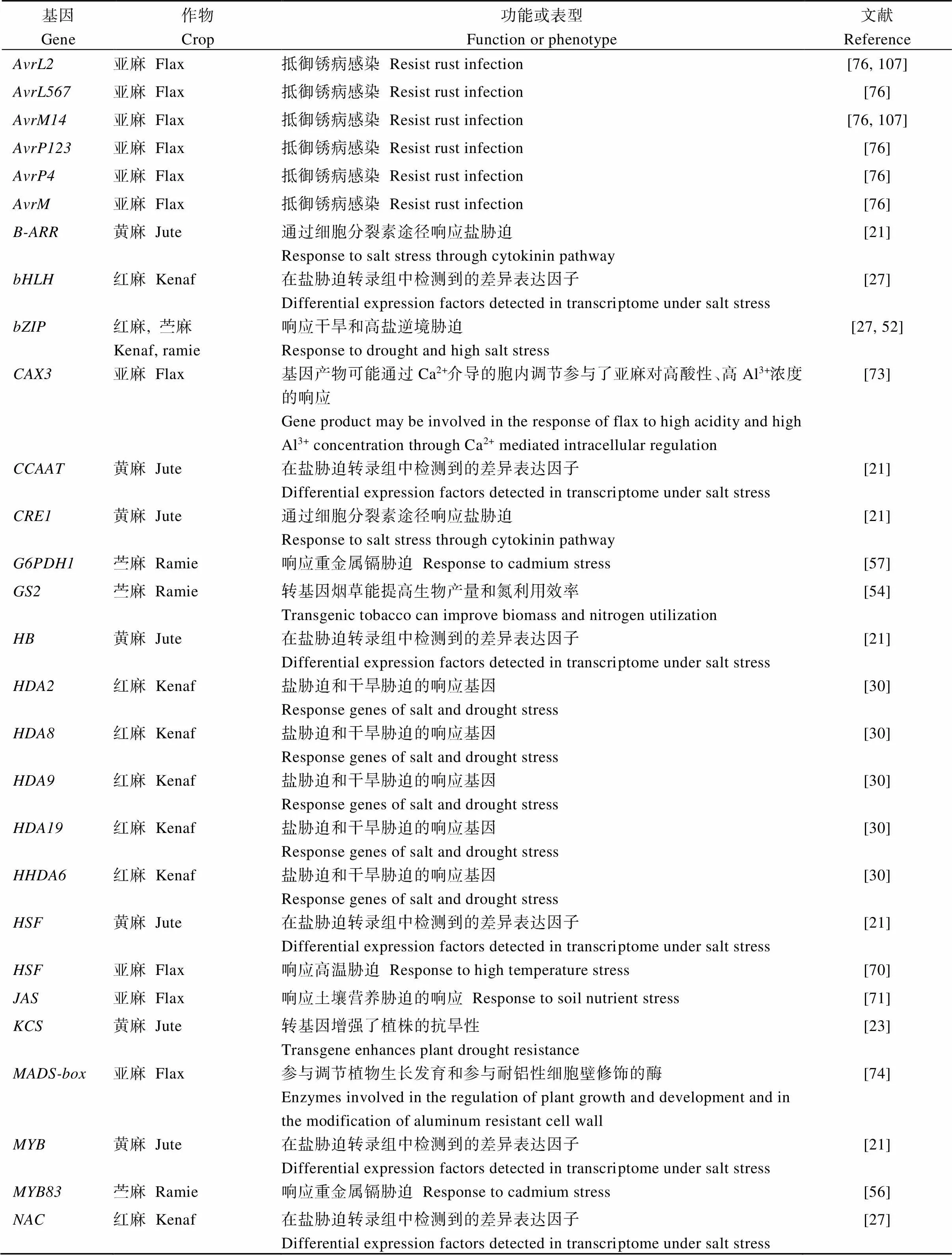
基因Gene作物Crop功能或表型Function or phenotype文献Reference AvrL2亚麻 Flax抵御锈病感染 Resist rust infection[76, 107] AvrL567亚麻 Flax抵御锈病感染 Resist rust infection[76] AvrM14亚麻 Flax抵御锈病感染 Resist rust infection[76, 107] AvrP123亚麻 Flax抵御锈病感染 Resist rust infection[76] AvrP4亚麻 Flax抵御锈病感染 Resist rust infection[76] AvrM亚麻Flax抵御锈病感染 Resist rust infection[76] B-ARR黄麻 Jute通过细胞分裂素途径响应盐胁迫Response to salt stress through cytokinin pathway[21] bHLH红麻 Kenaf在盐胁迫转录组中检测到的差异表达因子Differential expression factors detected in transcriptome under salt stress[27] bZIP红麻, 苎麻 Kenaf, ramie响应干旱和高盐逆境胁迫Response to drought and high salt stress[27, 52] CAX3亚麻 Flax基因产物可能通过Ca2+介导的胞内调节参与了亚麻对高酸性、高Al3+浓度的响应Gene product may be involved in the response of flax to high acidity and high Al3+ concentration through Ca2+ mediated intracellular regulation[73] CCAAT黄麻 Jute在盐胁迫转录组中检测到的差异表达因子Differential expression factors detected in transcriptome under salt stress[21] CRE1黄麻 Jute通过细胞分裂素途径响应盐胁迫Response to salt stress through cytokinin pathway[21] G6PDH1苎麻 Ramie响应重金属镉胁迫 Response to cadmium stress[57] GS2苎麻 Ramie转基因烟草能提高生物产量和氮利用效率 Transgenic tobacco can improve biomass and nitrogen utilization[54] HB黄麻 Jute在盐胁迫转录组中检测到的差异表达因子Differential expression factors detected in transcriptome under salt stress[21] HDA2红麻 Kenaf盐胁迫和干旱胁迫的响应基因Response genes of salt and drought stress[30] HDA8红麻 Kenaf盐胁迫和干旱胁迫的响应基因Response genes of salt and drought stress[30] HDA9红麻 Kenaf盐胁迫和干旱胁迫的响应基因Response genes of salt and drought stress[30] HDA19红麻 Kenaf盐胁迫和干旱胁迫的响应基因Response genes of salt and drought stress[30] HHDA6红麻 Kenaf盐胁迫和干旱胁迫的响应基因Response genes of salt and drought stress[30] HSF黄麻 Jute在盐胁迫转录组中检测到的差异表达因子Differential expression factors detected in transcriptome under salt stress[21] HSF亚麻 Flax响应高温胁迫 Response to high temperature stress[70] JAS亚麻 Flax响应土壤营养胁迫的响应 Response to soil nutrient stress[71] KCS黄麻 Jute转基因增强了植株的抗旱性Transgene enhances plant drought resistance[23] MADS-box亚麻 Flax参与调节植物生长发育和参与耐铝性细胞壁修饰的酶Enzymes involved in the regulation of plant growth and development and in the modification of aluminum resistant cell wall[74] MYB黄麻 Jute在盐胁迫转录组中检测到的差异表达因子Differential expression factors detected in transcriptome under salt stress[21] MYB83苎麻 Ramie响应重金属镉胁迫 Response to cadmium stress[56] NAC红麻 Kenaf在盐胁迫转录组中检测到的差异表达因子Differential expression factors detected in transcriptome under salt stress[27]
(续附表2)
附表3 主要麻类作物特异性状候选基因信息
Table S3 Information of candidate genes for specific traits in main bast fiber crops
(续附表3)
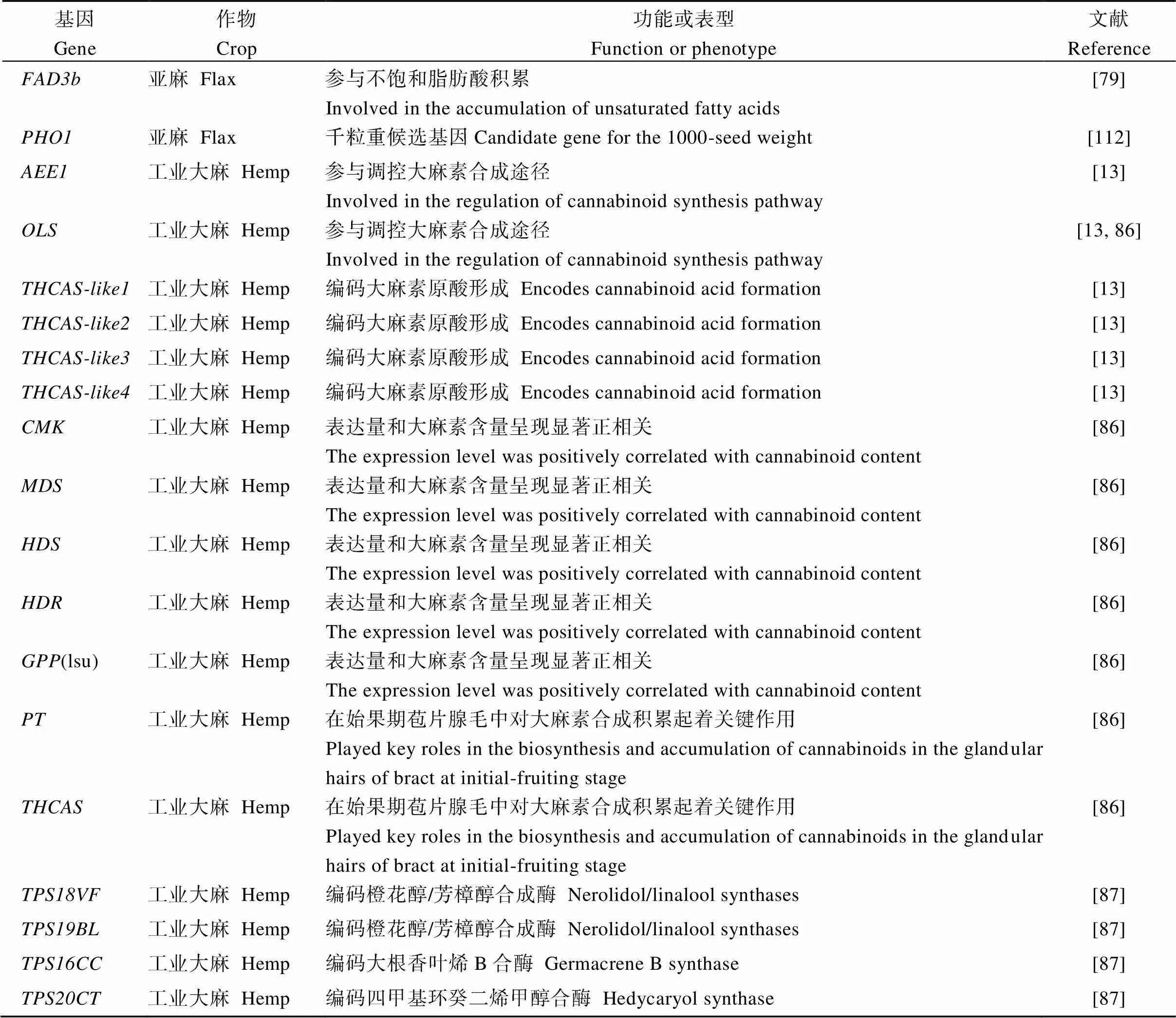
基因Gene作物Crop功能或表型Function or phenotype文献Reference FAD3b亚麻 Flax参与不饱和脂肪酸积累Involved in the accumulation of unsaturated fatty acids[79] PHO1亚麻 Flax千粒重候选基因Candidate gene for the 1000-seed weight[112] AEE1工业大麻 Hemp参与调控大麻素合成途径Involved in the regulation of cannabinoid synthesis pathway[13] OLS工业大麻 Hemp参与调控大麻素合成途径Involved in the regulation of cannabinoid synthesis pathway[13, 86] THCAS-like1工业大麻 Hemp编码大麻素原酸形成 Encodes cannabinoid acid formation[13] THCAS-like2工业大麻 Hemp编码大麻素原酸形成 Encodes cannabinoid acid formation[13] THCAS-like3工业大麻 Hemp编码大麻素原酸形成 Encodes cannabinoid acid formation[13] THCAS-like4工业大麻 Hemp编码大麻素原酸形成 Encodes cannabinoid acid formation[13] CMK工业大麻 Hemp表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content[86] MDS工业大麻 Hemp表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content[86] HDS工业大麻 Hemp表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content[86] HDR工业大麻 Hemp表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content[86] GPP(lsu)工业大麻 Hemp表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content[86] PT工业大麻 Hemp在始果期苞片腺毛中对大麻素合成积累起着关键作用 Played key roles in the biosynthesis and accumulation of cannabinoids in the glandular hairs of bract at initial-fruiting stage[86] THCAS工业大麻 Hemp在始果期苞片腺毛中对大麻素合成积累起着关键作用 Played key roles in the biosynthesis and accumulation of cannabinoids in the glandular hairs of bract at initial-fruiting stage[86] TPS18VF工业大麻 Hemp编码橙花醇/芳樟醇合成酶 Nerolidol/linalool synthases[87] TPS19BL工业大麻 Hemp编码橙花醇/芳樟醇合成酶Nerolidol/linalool synthases[87] TPS16CC工业大麻 Hemp编码大根香叶烯B合酶Germacrene B synthase[87] TPS20CT工业大麻 Hemp编码四甲基环癸二烯甲醇合酶 Hedycaryol synthase[87]
Genomics and genetic improvement in main bast fiber crops: advances and perspectives
XU Yi1,2,3, ZHANG Li-Lan1,2,3, QI Jian-Min1,2, ZHANG Lie-Mei1, and ZHANG Li-Wu1,2,3,*
1Key Laboratory of Ministry of Education for Genetics, Breeding and Multiple Utilization of Crops / Fujian Key Laboratory for Crop Breeding by Design / College of Agriculture, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China;2Experiment Station of Jute and Kenaf in Southeast China, Ministry of Agriculture and Rural Affairs / Public Platform of Fujian for Germplasm Resources of Bast Fiber Crops / Fujian International Science and Technology Cooperation Base for Genetics, Breeding and Multiple Utilization Development of Southern Economic Crops, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China;3Center for Genomics and Biotechnology, Haixia Institute of Science and Technology, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China
With the development of sequencing technology, main bast fiber crops (jute, kenaf, ramie, flax, and hemp) have completed genome sequencing from 2011 to 2020, which marks that the science of bast fiber crops has entered the era of genome. Firstly, this paper reviews the genome sequencing of bast fiber crops. Secondly, the important gene identification of bast fiber crops is also reported. Based on reference genome and transcriptome sequencing, a large number of candidate genes related to fiber development and response to abiotic stress have been detected, corresponding to the species characteristics of bast fiber and the adversity agriculture of “not competing with food”. Meanwhile, candidate genes for specific bast fiber crops have also been identified, such as male fertility in kenaf, seed oil content in flax, cannabinoid related candidate genes. Thirdly, the completion of bast fiber crop genome sequencing provides the possibility of omics-based genetic improvement, which will facilitate to study the formation of bast fiber and evolution mechanisms of bast fiber crop germplasms and systematically analyze the molecular basis for the formation of agronomic traits such as fiber yield, fiber quality, disease resistance, and stress tolerance. Also, it will facilitate to establish a high-throughput genotype-phenotype database, mine excellent gene resources, and create new germplasm. Moreover, it will facilitate to establish efficient rapid breeding technology systems by the innovation and combination of molecular marker-assisted selection, genome selection, transgenic technology and so on. To meet the market demand particularly bast fiber crop-related industries and adapt to the production model of bast fiber crops, we should breed new bast fiber crop varieties with high yield, high efficiency, stress resistance, disease resistance, suitable for light simplification and mechanization cultivation, high quality, and special purpose. Although the important information of gene resources and loci has been obtained from the reference genomes, there are still a series of challenges that how to utilize the existing resources efficiently for genetic improvement of bast fiber crops, such as stable and efficient genetic transformation system, construction of gene editing system, and genome selection breeding.
major bast fiber crops; genome; gene; genetic improvement
10.3724/SP.J.1006.2021.04121
本研究由国家自然科学基金项目(31771369, 31972968)和国家现代农业产业技术体系建设专项(CARS-19-E06)资助。
This study was supported by the National Natural Science Foundation of China (31771369, 31972968) and the China Agriculture Research System (CARS-19-E06).
张立武, E-mail: lwzhang@fafu.edu.cn; zhang_liwu@hotmail.com
E-mail: 1275924118@qq.com
2020-05-01;
2021-01-11;
2021-01-25.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20210125.1444.002.html
猜你喜欢
纺织报告(2022年8期)2022-11-21
特种经济动植物(2022年5期)2022-05-09
特种经济动植物(2021年11期)2021-11-14
雨花(2018年10期)2018-11-15
中国纤检(2018年10期)2018-10-29
散文诗世界(2017年3期)2017-11-13
家庭百事通·健康一点通(2016年7期)2016-08-04
纺织服装流行趋势展望(2016年6期)2016-05-04
纺织服装流行趋势展望(2016年2期)2016-05-04
西江月(2015年12期)2015-11-17
