
可降解聚羟基乙酸低聚物改性聚酯的合成及其性能
2021-04-06 05:21靳琳琳田俊凯李家炜戚栋明沈晓炜邬春涛
纺织学报 2021年1期
靳琳琳, 田俊凯, 李家炜, 戚栋明, 沈晓炜, 邬春涛
(1. 浙江理工大学 先进纺织材料与制备技术教育部重点实验室, 浙江 杭州 310018; 2. 浙江卫星石化股份有限公司, 浙江 嘉兴 314000; 3. 宁波市生态环境局, 浙江 宁波 315000)
石油基聚酯——聚对苯二甲酸乙二醇酯(PET)具有优异的耐热性、耐化学药品性和物理力学性能,且制备成本低,被广泛应用于化纤、薄膜、工程塑料等行业[1-2]。但PET材料具有较强的化学惰性,存在难以降解问题,是白色污染的主要污染源[3-4],因此,聚酯废料的降解与回收利用已成为当前聚酯工业的重点课题[5-6]。
脂肪族聚酯,如聚乳酸(PLA)、聚羟基乙酸(PGA)、聚丁二酸丁二醇酯(PBS)等被认为是具有代表性的生物降解聚合物[5]。近年来,研究人员通过与脂肪族聚酯共混、共聚改性PET材料,可有效提高聚酯材料的降解性能。例如,高翠丽等[7]以PLA与PET共混挤出的方法制备PET/PLA共混物,其中PET和PLA共混比例为9∶1时共混物的降解率最大。Olewnik等[8]采用熔融缩聚法制备了新型可生物降解聚对苯二甲酸乙二醇酯-co-聚乙醇酸共聚酯(PET-co-PGA),具有优良的降解性能。然而,直接熔融缩聚所合成的共聚物的分子质量较低,力学性能较差,限制了其在纤维材料领域的应用。
固相缩聚是提高聚酯相对分子质量的一种有效方法,也是工业上制备工业级PET聚酯的重要方法[9-10]。如果能利用熔融缩聚和固相缩聚联用的工艺合成 PET-co-PGA共聚酯,有望获得降解性能与力学性能俱佳的改性聚酯,因此,本文采用该联用工艺路线合成一系列PET-co-PGA共聚酯,研究了PGA的加入对改性聚酯的热性能、结晶行为、力学性能以及降解性能的影响,并对其作用机制进行分析。
1 实验部分
1.1 原 料
对苯二甲酸(PTA),中国石化仪征化纤股份有限公司;乙二醇(EG)、三氧化二锑(Sb2O3)、多聚磷酸、羟基乙酸(GA)、苯酚、1,1,2,2-四氯乙烷、磷酸二氢钾(KH2PO4)和磷酸氢钠(Na2HPO4),均为分析纯,上海国药集团化学试剂有限公司。
1.2 共聚酯的合成
首先羟基乙酸(GA)发生缩聚反应,合成PGA低聚物;然后PTA与EG发生酯化反应,生成对苯二甲酸乙醇酯(BHET);最后,采用熔融缩聚和固相缩聚联用工艺,PET中的羧基与羟基乙酸中的羟基发生酯化反应产生新型可降解PGA改性聚酯,反应机制如图1所示。
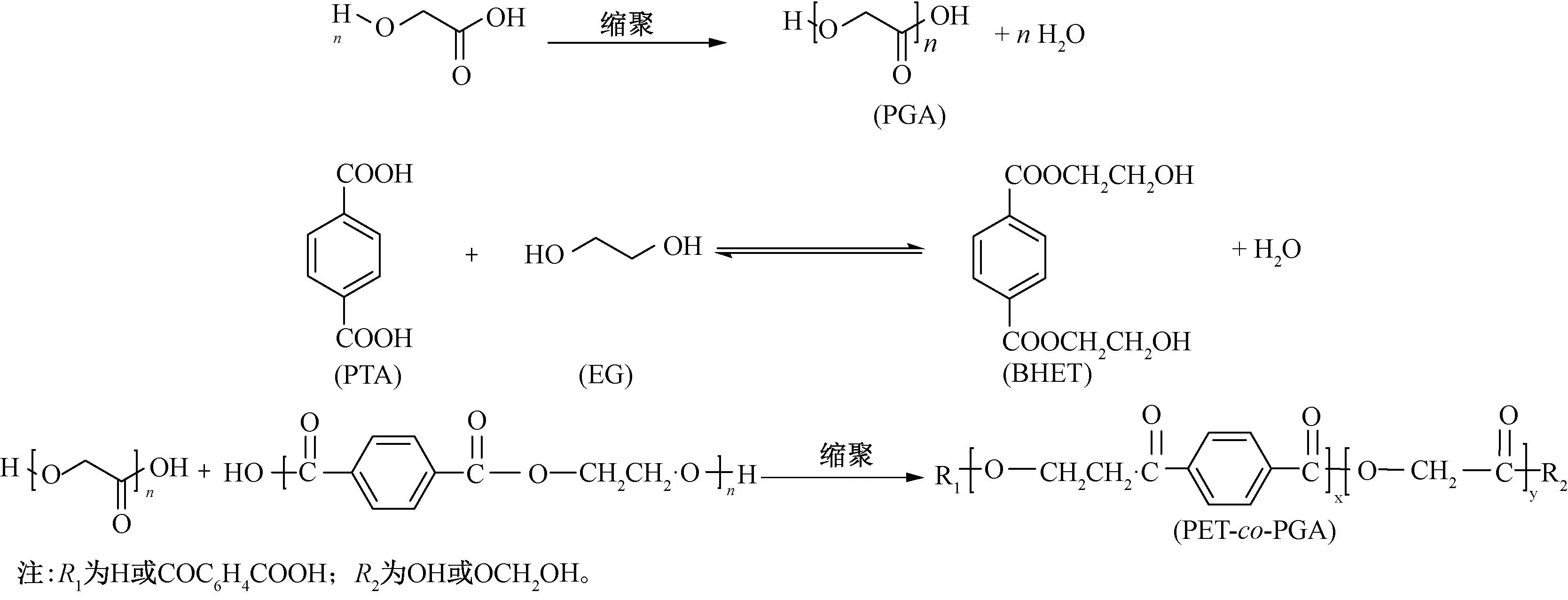
图1 聚羟基乙酸低聚物改性聚酯的合成路线
1.2.1 PGA低聚物的合成
将250.0 g羟基乙酸加入带搅拌的500 mL的四口烧瓶中,加热完全熔融后,缓慢抽真空,在(190±2) ℃温度下保持绝对压力为81 kPa,缩聚反应20 min,制得PGA低聚物[11]。
1.2.2 熔融缩聚
取149.4 g对苯二甲酸,74.4 g乙二醇,0.045 g三氧化二锑和0.035 g多聚磷酸,加入到1 L的聚酯反应釜(扬州普利特有限公司)中,在470 kPa、230 ℃环境下保持2 h,分馏出水分。然后,将不同质量分数的PGA投入到反应釜中进行缩聚反应,缩聚温度为265 ℃,保持绝对压力小于100 Pa,反应3 h后停止反应出料。合成共聚酯的原材料配比如表1所示。
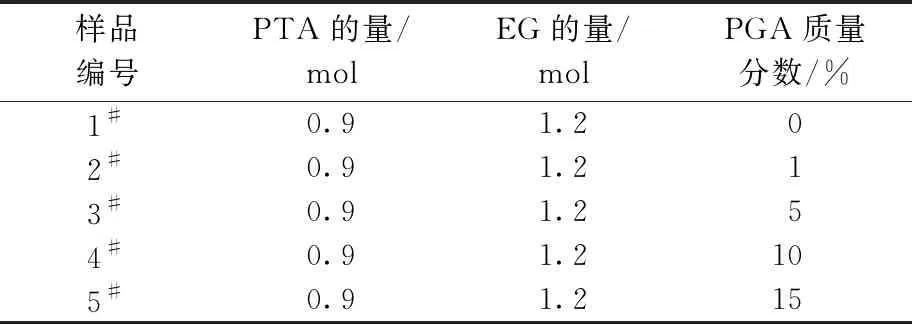
表1 聚羟基乙酸低聚物改性聚酯投料比
1.2.3 固相缩聚
取100 g熔融缩聚得到的共聚酯加入四口烧瓶中,在温度为200 ℃,绝对压力为100 Pa条件下固相缩聚反应35 h,完成固相缩聚得到PET-co-PGA共聚酯。
1.3 改性聚酯纤维膜的制备
利用静电纺丝装置,在外加电压为15 kV、喷丝头与接收装置间的距离为15 cm、纺丝溶液的流速为0.5 mL/h的条件下制备出共聚酯纳米纤维膜。
1.4 性能测试与表征
1.4.1 化学结构测试
采用AVANCE III型核磁共振谱仪(德国Bruker公司),以氘代三氟乙酸为溶剂,测定PET-co-PGA共聚酯的化学结构。
1.4.2 特征黏度测试
准确称量(0.128±0.005) g共聚酯试样加入锥形瓶中,并加入25 mL苯酚-1,1,2,2-四氯乙烷(二者质量比为1∶1)混合溶剂,于80 ℃恒温溶解,冷却至室温。然后,用乙醇反复洗涤干燥过的玻璃砂芯漏斗过滤,除去聚合过程中可能混入的杂质。然后取过滤后的溶液加入PXWSN-4B型乌氏黏度计(上海平轩科学仪器有限公司)中,将乌氏黏度计置于(25±0.05) ℃的恒温水浴中测试。增比黏度ηnp计算公式为
式中:tn为溶液流经时间,s;t0为空白溶剂的流经时间,s。
特性黏度 [η]计算公式为
式中:c为聚合物溶液的浓度,mol/L。
1.4.3 热学性能测试
利用Discovery 25型差示扫描量热仪(美国TA公司)以10 ℃/min的升温速率从50 ℃升温至260 ℃,并保持5.0 min以消除样品热历史,然后以10 ℃/min的速率降温至50 ℃,保持5 min,再以10 ℃/min升温至260 ℃。
1.4.4 拉伸性能测试
采用DHG-9070 A型鼓风干燥箱(上海一恒科学仪器有限公司)烘干共聚酯,然后用minijet II实验注塑机(德国haake公司)制备共聚酯的实验样条,厚度为1.68 cm。然后,根据ASTM D638—2010《塑料拉伸性能的标准试验方法》,采用AGS-10KND型精密万能试验机(日本岛津公司)测试样品的拉伸力学性能。拉伸速率为100 mm/min,每组样品测试5次,取平均值。
1.4.5 降解性能测试
称取1.18 g KH2PO4和4.30 g Na2HPO4在1 000 mL的容量瓶中配制成pH值为10.01的碱性缓冲溶液。用筛子筛选出粒径为150~178 μm的颗粒,准确称取试样质量a为0.50 g,加入到带塞子的试管中,在恒温(37 ℃)水浴下测试样品的保留率和降解率,测试pH值为10.01的碱性体系。测试1个周期(7 d)后过滤,真空干燥24 h,准确称量剩余质量记为b(g)。利用下式分别计算降解率(D)和保留率(F):
1.4.6 表面形貌观察
将所得纤维膜裁剪成规格为1 cm × 1 cm的正方形,用导电胶将其贴在样品台上进行喷金处理,采用JSM-5610LV型扫描电子显微镜(SEM,日本电子株式会社)观察其表面形貌,加速电压为3 kV。
1.4.7 荧光显微降解性能测试
取100.0 g PET-co-PGA共聚酯,加入5.0 mg的五甲基氟化硼二吡咯荧光化合物(BODIPY),溶解在六氟异丙醇(HFIP)溶液中配成浓溶液,在10 kV的电压下静电喷丝。利用ECLIPSE Ti-S型倒置荧光显微镜(日本Nikon公司)对降解前和在pH值为10.01的碱性体系中降解6周后的聚酯纤维进行紫外激发,并拍摄其荧光照片。
2 结果与讨论
2.1 化学结构分析
PET-co-PGA共聚酯的核磁共振氢谱(1H-NMR)图如图2所示。可知,曲线在化学位移δ位于5.26、4.79处为与酯基相连的—CH2—上氢原子的质子峰,δ位于4.38处为与羟基相连的—CH2—上氢原子的质子峰,δ位于5.00 处为乙二醇中氢质子峰,δ位于8.32处为苯环上氢原子的质子峰,δ位于11.50处为溶剂CF3COOD中质子吸收峰。实验结果说明核磁共振氢谱质子峰归属与预期结构一致。

图2 PET-co-PGA改性聚酯的化学结构式和核磁共振氢谱图
2.2 特性黏度分析
经特性黏度测试,纯PET试样(1#)的特性黏度值为1.03 dL/g,添加PGA质量分数为1%、5%、10%和15%时,试样的特性黏度分别为1.05、1.09、1.06、和1.02 dL/g,PET-co-PGA共聚酯的特性黏值在1.02~1.09 dL/g之间变化,表明所合成的纯PET和PET-co-PGA共聚酯的特性黏度在同一数量级,所有样品的特性黏度值均在1.0 dL/g以上,达到了商业化聚酯的要求[13]。
2.3 共聚酯的热性能及结晶行为分析
为探究PGA低聚物对共聚酯热性能和结晶行为的影响,对纯PET和PET-co-PGA共聚酯进行DSC测试,结果如图3所示。相应的数据如表2所示。由图3(a)可知,随着PGA质量分数的增加,PET-co-PGA共聚酯的玻璃化转变温度(Tg)逐渐降低。当PGA添加质量分数为5%时,共聚酯(样品3#)的Tg由纯PET(样品1#)的82 ℃下降至72 ℃,与纯 PET相比下降了12%,表明柔性脂肪族PGA的引入,增强了聚酯大分子链的柔顺性,大分子链运动更加容易。
从图3(b)可看出,随着柔性脂肪族PGA质量分数的增加,PET-co-PGA共聚酯的结晶温度(Tc)逐渐降低,甚至消失。当PGA质量分数为5%时,样品3#的结晶温度由纯PET的180 ℃下降到140 ℃,且峰形变得宽矮不对称。当PGA质量分数大于10%时,样品4#和5#的冷结晶峰完全消失。这说明柔性PGA的加入提高了共聚酯结晶速率。另一方面,根据聚合物结晶原理,可通过结晶温度与玻璃化转变温度之差(Tc-Tg)来评价其在变温过程中的结晶难易程度,差值越大表示越难结晶[14]。由表2可知,随PGA质量分数增加,Tc-Tg差值越来越小,表明结晶变得越来越容易。
由图3(c)可看到,随着PGA质量分数的增加,PET-co-PGA共聚酯的熔融峰朝着低温方向移动,熔融峰形变宽矮,Tm逐渐降低。当PGA添加质量分数为1%时,样品2#的Tm由纯PET的248 ℃下降至239 ℃,相应的熔融焓(ΔHm)由67.57 J/g略微增加至69.63 J/g,其结晶度也略微增加至58.9%,这主要是添加的少量的PGA起到异相成核作用。当添加质量分数为5%的PGA时,样品3#的Tm由纯PET的248 ℃下降至218 ℃,相应的熔融焓(ΔHm)由67.57 J/g降至57.10 J/g。这可归因于PGA的加入使共聚酯主链的规整性降低,不易排列形成高度有序的晶格,导致熔点下降[12]。
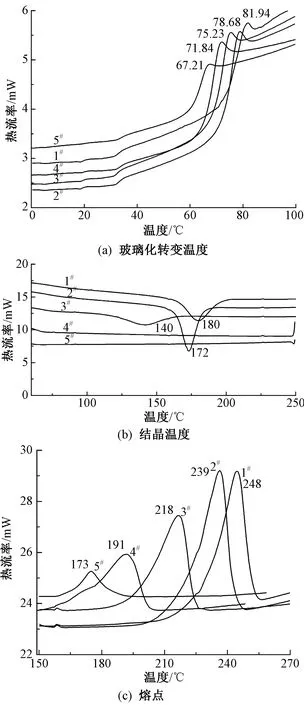
图3 纯PET和PET-co-PGA共聚酯的DSC曲线

表2 纯PET和PET-co-PGA共聚酯的DSC数据
综上所述,因为柔性脂肪族PGA链的加入使得聚酯的规整性被打破,所以PET-co-PGA共聚酯的Tg、Tm和结晶能力均随着PGA低聚物质量分数的增加而减小。
2.4 力学性能分析
纯PET和PET-co-PGA共聚酯的拉伸性能测试结果如表3所示。与纯PET(1#)相比,样品2#和3#断裂强度和弹性模量略微增加。值得注意的是,样品3#的断裂伸长率由样品1#的13.09%左右增加至15.28%左右,较纯PET提升了14.6%,说明了试样3#较纯PET试样表现出韧性特征。这是由于加入柔性脂肪族的PGA在PET基体形成稳定的PGA橡胶相,增强了PGA相与PET基体的界面相容性,导致共聚酯韧性增加;但随着PGA质量分数的增加,共聚物的断裂强度、弹性模量、断裂伸长率均呈现明显的下降趋势,力学性能变差。这可归因于随PGA加入量的增加,降低了结晶性能,使得链段间的相互作用减弱。因此,在PGA质量分数为5%时,芳香-脂肪共聚酯的柔性链段保持了PET基体的高拉伸强度,同时增加了材料的韧性[12]。
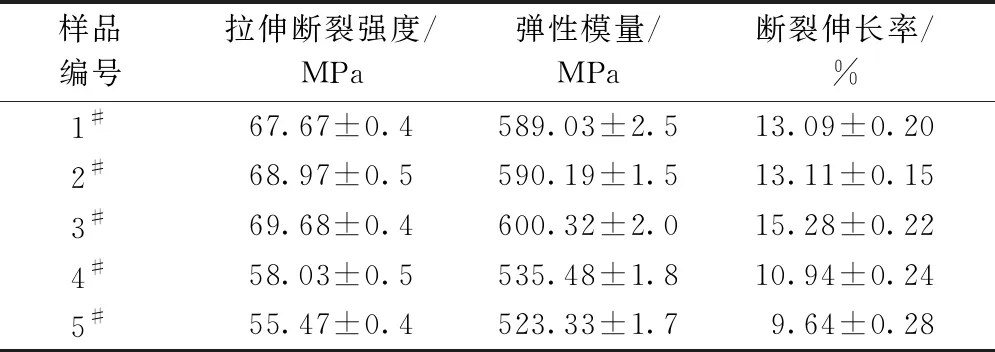
表3 PET-co-PGA共聚酯的力学性能
2.5 降解性能分析
图4示出纯PET和PET-co-PGA共聚酯在pH值为10.01缓冲溶液中的降解行为[15]。可知,降解12周后,样品1#、2#、3#、4#的PET-co-PGA共聚酯保留率分别为98.5%、88.0%、84.6%和79.5%,说明随着脂肪族PGA低聚物质量分数的增加,共聚酯降解明显增加。这是由于脂肪族的PGA低聚物加入提升了共聚酯大分子链的运动能力,分子链更易被水解为低分量的低聚物,加快了降解速率。这一结果与共聚酯的Tg值相一致[16]。
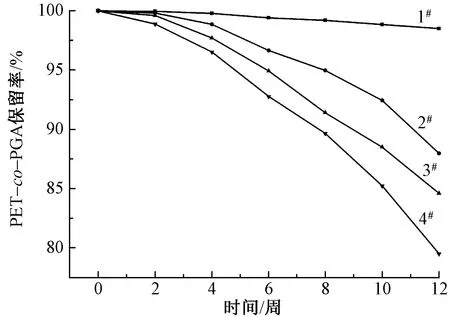
图4 共聚酯碱性条件下的降解行为
对样品4#降解前后的纳米纤维膜进行SEM和荧光显微镜观察,结果如图5、6所示。

图5 共聚酯纤维降解过程的扫描电镜照片(×5 000)
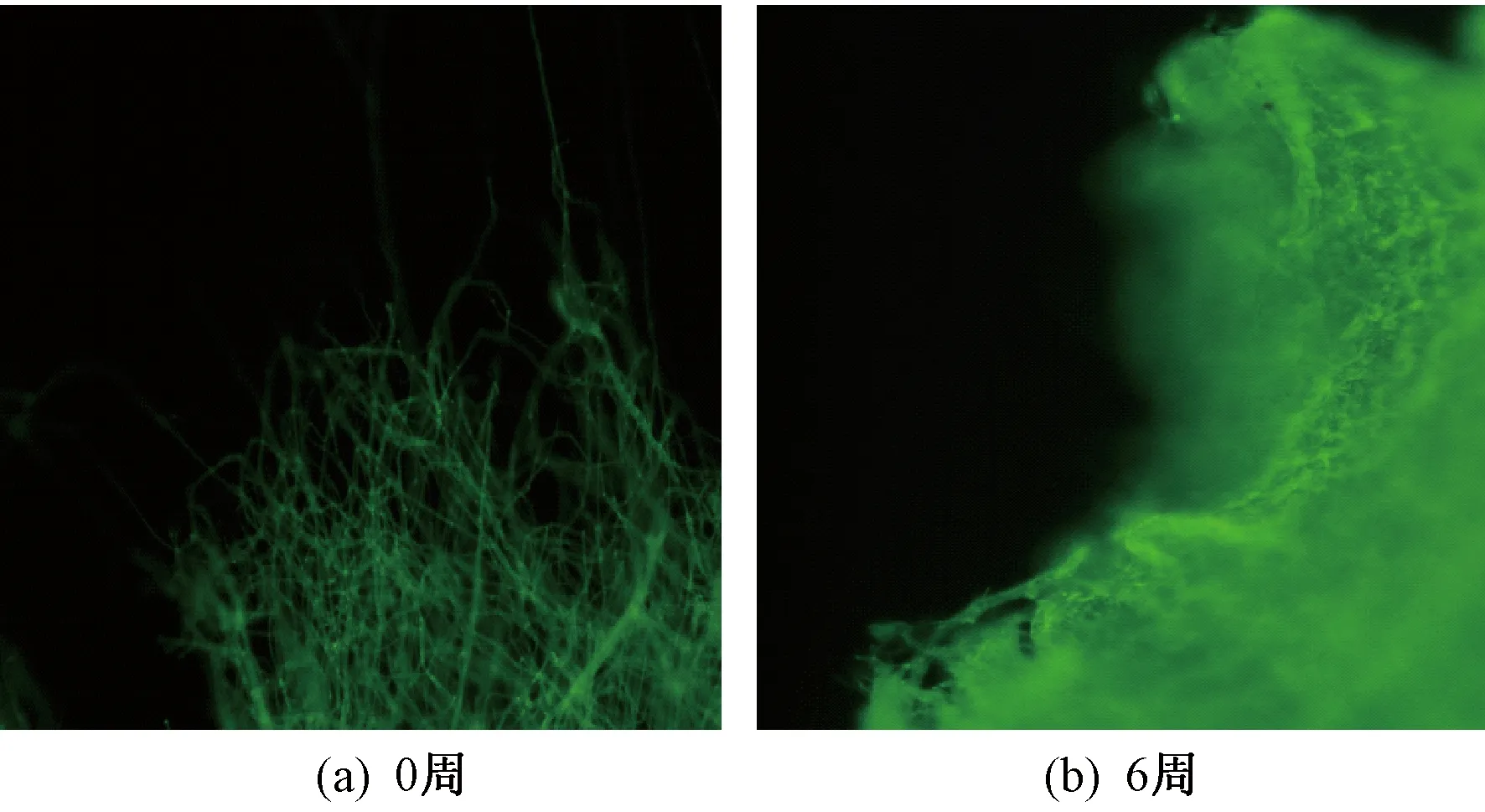
图6 共聚酯纤维在降解过程中结构形态变化的荧光照片
由图5可看出,降解前纤维呈表面光滑的圆柱形,纤维直径分布均匀,降解6周后纤维表面出现不同程度的褶皱和粗糙现象。与此同时,由图6可知,降解前纤维表面为层次分明的丝状,降解6周后纤维重现雾状,观察不出明显的纤维状结构,说明共聚酯纳米纤维膜发生明显降解。上述结果表明,加入生物基脂肪族PGA低聚物,提升了改性聚酯降解速率。
3 结 论
1)通过熔融缩聚和固相缩聚工艺路线,成功制备了PGA改性PET共聚酯,通过核磁氢谱测试表明,PET-co-PGA共聚酯的分子结构与预期化学结构相吻合,共聚酯样品的特性黏度值均大于1 dL/g,达到商业化聚酯水平。
2)PET-co-PGA共聚酯的玻璃化转变温度、熔点和结晶能力随着PGA低聚物占比增加而降低,说明聚酯大分子的规整性被打破。虽然聚酯分子链上加入了柔性的脂肪族PGA链段,但力学性能测试发现在低PGA质量分数共聚酯的断裂伸长率优于纯PET,表现出韧性。
3)PET-co-PGA共聚酯在实验条件下均可降解,其中降解速率随着PGA质量分数增加而加快,而纯PET试样在实验条件下无明显降解。
猜你喜欢
纺织学报(2022年3期)2022-03-28
润滑油(2021年5期)2021-11-23
疯狂英语·新读写(2021年5期)2021-11-23
纺织科学研究(2021年1期)2021-03-19
金桥(2020年10期)2020-11-26
润滑油(2019年6期)2020-01-01
中国新技术新产品(2018年17期)2018-11-29
爱你(2017年2期)2017-11-24
食品与生活(2016年5期)2016-05-23
养生保健指南(2016年9期)2016-05-14
