
高效液相色谱法测定虾青素油中虾青素的含量
2021-04-01 03:26孔凡华徐佳佳郭倩白沙沙李东方从容邱楠楠崔亚娟
食品与发酵工业 2021年6期
孔凡华,徐佳佳,郭倩,白沙沙,李东,方从容,邱楠楠*,崔亚娟*
1(国家食品安全风险评估中心,国家卫生健康委员会食品安全风险评估重点实验室,北京,100021) 2(北京市营养源研究所,北京,100069)
虾青素为萜烯类不饱和化合物,化学名称是3,3′-二羟基-4,4′-二酮基-β,β′-胡萝卜素,是一种具有超级抗氧化活性的类胡萝卜素[1],其抗氧化性能是维生素E、维生素C、β-胡萝卜素、叶黄素和玉米黄质的几十倍甚至几百倍[2-4]。雨生红球藻、红法夫酵母、南极磷虾、一些浮游生物、藻类、螃蟹外壳、河螯虾外壳、牡蛎、三文鱼皮、鲑鱼等动物中含有虾青素。虾青素具有多种生物功能,它能终止生物系统中炎症的发生[5]、对抗自由基损伤以维持免疫系统防御[6]、降低心脏病发病的风险[7]、减轻神经退行性疾病带来的影响[8]、抑制肿瘤发生和生长[9]及对抗紫外线引起的光氧化损伤[10]等。
虾青素具有较长的共轭双键链,结构复杂,化学性质不稳定,存在多种同分异构体,虾青素及其异构体的定性定量分析是结构解析和功能研究的基础。天然虾青素大多与脂肪酸结合,以酯化的形态存在,但由于缺乏虾青素酯标准品,且人工合成虾青素酯困难,限制了虾青素酯的定性定量研究。虾青素酯脱去脂肪酸链后可以形成游离的虾青素,包括全反式虾青素、9-顺-虾青素、13-顺-虾青素等,使虾青素的定量研究变得容易操作[11]。目前,虾青素脱脂的主要方法是碱性皂化和酶解方法,碱性皂化脱脂会产生副产物并引起虾青素降解[12],食品化学法典(FCC)(2012)[13]采用来自假单胞杆菌的胆固醇酯酶(EC 3.1.1.13)进行虾青素酯的酶解,可以使虾青素酯水解完全。色谱技术的发展在虾青素的定性定量分析上起了非常重要的作用,随着紫外可见光谱、质谱、核磁共振波谱法、红外光谱、拉曼光谱和圆二色光谱的快速发展,虾青素的结构鉴定变得更为简单容易,考虑到设备的成本,样品制备的复杂程度和方法的局限性,虾青素的定量分析方法主要是分光光度法和高效液相色谱法。分光光度法具有快速简单、适用范围广、成本低等优点,但是只能对虾青素总量进行估算,无法对其中虾青素的存在形式进行准确的测定。高效液相色谱法分离效果好、灵敏度高、选择性好成为目前测定虾青素的常用方法。我国测定虾青素的标准方法有《红球藻中虾青素的测定 液相色谱法》(GB/T 31520—2015)[14]适用于雨生红球藻藻粉中虾青素含量的测定;《水产品及其制品中虾青素含量的测定 高效液相色谱法》(SC/T 3053—2019)[15]适用于鱼类、甲壳类及虾粉、磷虾油等制品中虾青索含量的测定;《进出口动物源性食品中角黄素、虾青素的检测方法》(SN/T 2327—2009)[16]适用于黄鱼、鳗鱼、鸡肉、鸡蛋、鸭肝、猪肾和牛奶中角黄素、虾青素的测定与确证;《植物提取物 虾青素油》(T/CCCMHPIE 1.23—2016)[17]适用于以人工培养的红球藻属雨生红球藻为原料经提取精制后,得到的虾青素油中虾青素的测定。以上标准方法通过有机溶剂直接提取或者碱性皂化脱脂后提取虾青素,容易引起虾青素的降解[12],导致测量结果不准确或对测定结果造成干扰;标准方法多采用三相洗脱系统分离虾青素及其异构体,对液相系统要求高。本文参照虾青素检测的标准方法[14-17]和文献方法[13,18-23],设计单因素实验,对提取试剂、胆固醇酯酶酶解时间进行优化,以虾青素含量的高低作为评价指标,筛选最佳提取试剂和酶解条件;通过比较色谱柱、流动相等高效液相色谱条件,以全反式虾青素、9-顺-虾青素、13-顺-虾青素的分离度和峰形对称性作为评价指标,筛选最佳色谱条件,实现虾青素油中虾青素的有效提取和高效分离。以虾青素油为研究对象,进行方法学验证,考察方法的科学性、准确性和适用性,并比较实验建立的酶解方法与文献中的碱性皂化方法[18]对虾青素油中虾青素的提取效率,以期建立虾青素油中虾青素的精准分析检测技术。
1 材料与方法
1.1 材料与试剂
超临界二氧化碳萃取的雨生红球藻虾青素油,市购;全反式虾青素对照品(纯度96.9%),Dr.Ehrenstorfer GmbH;9-顺-虾青素、13-顺-虾青素对照品(纯度≥95.0%),Sigma公司;胆固醇酯酶16.3 U/mg,月旭科技股份有限公司。
甲醇、乙腈、甲基叔丁基醚、二氯甲烷、乙酸乙酯(均为色谱纯),美国Fisher公司;无水乙醇、无水硫酸钠、氢氧化钠、浓盐酸(分析纯),北京化工厂;石油醚(分析纯),福晨(天津)化学试剂有限公司;丙酮(分析纯),国药集团化学试剂有限公司;三羟甲基氨基甲烷(Tris)(分析纯),浙江联硕生物科技有限公司。
1.2 仪器与设备
BS224S分析天平,德国赛多利斯科学仪器(北京)有限公司;METTLER TOLEDO pH计,梅特勒-托利多国际贸易(上海)有限公司;QL-901斡旋振荡器,海门市其林贝尔仪器制造有限公司;ZHWY-110X30往复式恒温水浴摇床,上海智诚分析仪器制造有限公司;KQ-500DE型数控超声波清洗器,昆山市超市仪器有限公司;HITACHI CF16RXII 高速冷冻离心机,日本日立科技有限公司;安捷伦1260高效液相色谱仪,配二极管阵列检测器和ChemStation for LC systems色谱工作站,美国Agilent公司;YMCTMC30色谱柱,5 μm,250 mm×4.6 mm(内径)。
1.3 方法
1.3.1 溶液的配制
(1)0.021 mol/L氢氧化钠-甲醇溶液:称取0.084 g氢氧化钠,加入80 mL 甲醇溶解并定容到100 mL。
(2)0.05 mol/L Tris-HCl缓冲液(pH=7):准确称取3.028 5 g Tris,加入450 mL去离子水溶解后,用0.5 mol/L的盐酸调节pH至7.0,然后用去离子水定容至500 mL。
(3)胆固醇酯酶溶液:称取6.2 mg胆固醇酯酶于25 mL容量瓶中,用0.05 mol/L Tris-HCl缓冲液定容至刻度,胆固醇酯酶溶液浓度约为4 U/mL,现用现配。
(4)全反式虾青素、9-顺-虾青素、13-顺-虾青素标准储备液:分别准确称取全反式虾青素、9-顺-虾青素、13-顺-虾青素标准品1 mg,分别置于25 mL棕色容量瓶中,加丙酮溶解并定容至刻度,摇匀,得到浓度为40 μg/mL 标准储备液,现用现配。
(5)全反式虾青素、9-顺-虾青素、13-顺-虾青素混合标准工作溶液:准确移取全反式虾青素、9-顺-虾青素、13-顺-虾青素标准储备溶液,用丙酮稀释成各浓度均为0.80 μg/mL的标准工作溶液,用于定性,现用现配。
(6)全反式虾青素标准工作溶液:准确移取全反式虾青素标准储备溶液,用丙酮稀释成浓度分别为0.10、0.20、0.40、0.80、1.60、4.00 μg/mL的标准工作溶液,现用现配。
1.3.2 色谱条件的选择
孙伟红等[19]研究表明,C30色谱柱对虾青素异构体的分离效果和响应值优于C18色谱柱,更有利于定量结果的分析,所以本实验选择YMCTMC30色谱柱。参考标准方法和文献方法,选择甲基叔丁基醚-乙腈(体积比95∶5)[20]、甲醇-甲基叔丁基醚-水(体积比83∶15∶2)[21]、水-甲醇-二氯甲烷-乙腈(体积比4.5∶28∶22∶45.5)[22]、水-甲醇-二氯甲烷-乙腈(5∶85∶5∶5)[23]和甲醇-甲基叔丁基醚[24]等流动相体系洗脱,以全反式虾青素、9-顺-虾青素、13-顺-虾青素的分离度作为评价指标,优化全反式虾青素、9-顺-虾青素、13-顺-虾青素分离的色谱条件。
1.3.3 提取溶剂的选择
虾青素油用丙酮稀释500倍作为实验用虾青素油样品。实验参照食品化学法典FCCth(2012)[23]的方法,并加以改进。称取10 mg虾青素油样品于15 mL离心管中,样品称取15份,分别加入2 mL Tris-HCl缓冲液(0.05 mol/L,pH=7)和1 mL胆固醇酯酶(4 U/mL)溶液振荡均匀后,在37 ℃恒温水浴振荡器内振荡酶解60 min,取出立即冷却至室温。向每份酶解液中分别加入 2.0 mL石油醚、二氯甲烷-甲醇(体积比1∶3)、丙酮、乙腈、甲醇-乙酸乙酯-石油醚(体积比1∶1∶1),每种提取溶剂做3个平行,每份样品均加入1 g无水硫酸钠后在涡旋振荡器上涡旋提取2 min,静置分层后,分别将上层溶液转移至10 mL容量瓶中,重复提取2次,合并提取液于容量瓶中,氮气吹干后,用甲醇定容至刻度,溶液过0.45 μm有机系滤膜后,待测定。
1.3.4 酶解时间的选择
称取10 mg虾青素油样品于15 mL离心管中,分别加入2 mL Tris-HCl缓冲液(0.05 mol/L,pH=7)和1.0 mL胆固醇酯酶(4 U/mL)溶液振荡均匀后,在37 ℃ 恒温水浴振荡器内分别振荡酶解15、30、45、60、75、90、120 min,取出立即冷却至室温,用石油醚提取,其他操作步骤同1.3.3。
1.3.5 方法验证
按照GB/T 27404—2008《实验室质量控制规范 食品理化检测》[25]方法学要求,建立校准曲线及校准曲线的工作范围;逐步稀释样品上机测定,R(S/N)=3时的测定浓度作为检出限,R(S/N)=10时的测定浓度作为定量限;称取虾青素油样品,按照实验优化的方法,平行测定6次,计算虾青素的含量,并计算相对标准偏差,考察方法的重现性;根据虾青素油样品中虾青素的含量,按本底值的0.5倍、1倍、1.5倍3个浓度水平,进行3水平6平行加标回收实验,以回收率的高低评价方法的准确度。
1.3.6 碱性皂化脱脂方法
参见YUAN等[18]和苏芳[26]的方法,称取10 mg虾青素油样品于15 mL离心管中,加入5 mL 0.021 mol/L 氢氧化钠-甲醇溶液,5 ℃水浴中避光皂化60 min,加入 2 mL 纯净水和 2 mL 石油醚,在涡旋振荡器上振荡2 min,静置分层后,将上层溶液转移至10 mL 容量瓶中,重复提取2次至石油醚层无色,合并提取液于容量瓶中,氮气吹干后,用甲醇定容至刻度,溶液过0.45 μm有机系滤膜后,测定。
1.4 虾青素含量的计算
1.4.1 标准曲线的制作
将各浓度全反式虾青素标准品工作溶液,9-顺式虾青素和13-顺式虾青素标准品工作溶液按实验优化的色谱条件进行液相色谱分析,根据虾青素异构体组分的保留时间定性。选定峰面积相近的全反式虾青素的标准工作液多点校准定量,试样测定结果以3种虾青素同分异构体的总和计,外标法定量,同时,标准工作液和样液的响应值均应在仪器检测的线性范围内。
1.4.2 结果计算
试样中虾青素的含量X以质量分数计,按式(1)计算。
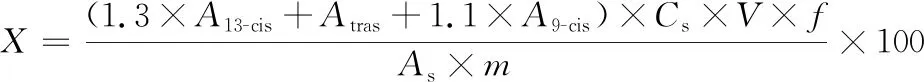
(1)
式中:X,试样中虾青素的含量,%;1.3,13顺-虾青素对全反式虾青素的校正因子;A13-cis,试样溶液中13-顺-虾青素的峰面积;Atras,试样溶液中全反式虾青素的峰面积;1.1,9-顺-虾青素对全反式虾青素的校正因子;A9-cis,试样溶液中9-顺-虾青素的峰面积;Cs,标准工作液中全反式虾青素的含量,μg/mL;V,试样溶液体积,mL;As,标准工作液中全反式虾青素的峰面积;m,试样质量,mg;f,稀释倍数。
1.5 数据处理
通过与仪器配套的 ChemStation for LC systems工作站软件完成数据的采集与处理。
数据的差异性分析在SPSS软件上进行,P<0.05时,说明差异水平显著。
2 结果与分析
2.1 色谱条件的选择
不同流动相体系对全反式虾青素、9-顺-虾青素、13-顺-虾青素分离效果见表1。
目前,多采用三相洗脱系统[14-15,17,19,27]或者正相洗脱系统[26]分离虾青素及其异构体,对液相系统要求较高,且普适性较差。本实验通过优化液相色谱条件,采用甲醇-甲基叔丁基醚两相梯度洗脱,可以实现3种虾青素异构体的有效分离,结果如图1所示,优化得到分离虾青素的色谱条件为:
色谱柱:YMCTMC30色谱柱,5 μm,250 mm×4.6 mm(内径)或相当者;柱温:30 ℃;流动相:见表1;流速:1.0 mL/min;检测波长:472 nm;进样量:20 μL。
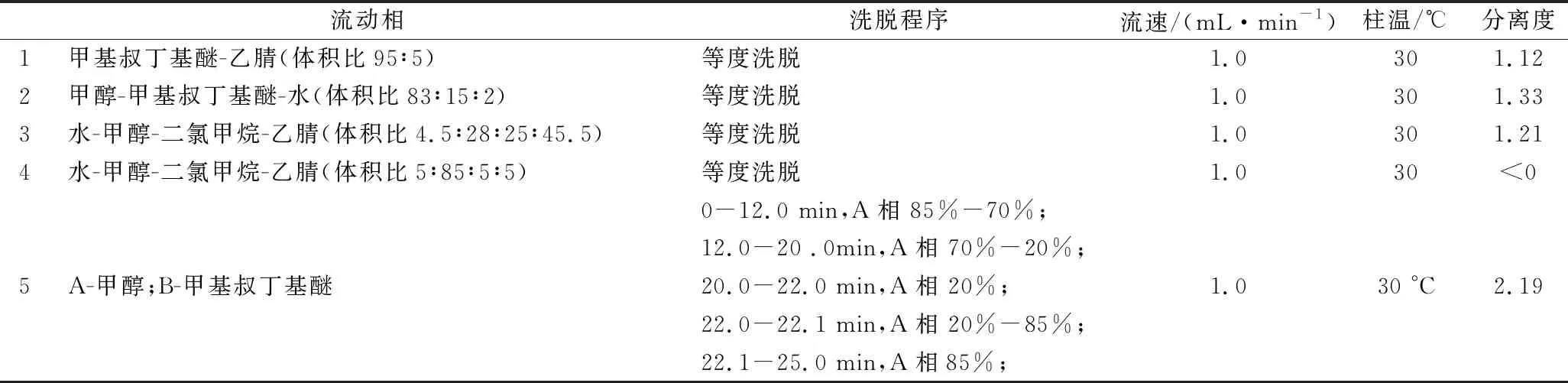
表1 色谱条件的优化Table 1 Optimization of chromatographic conditions
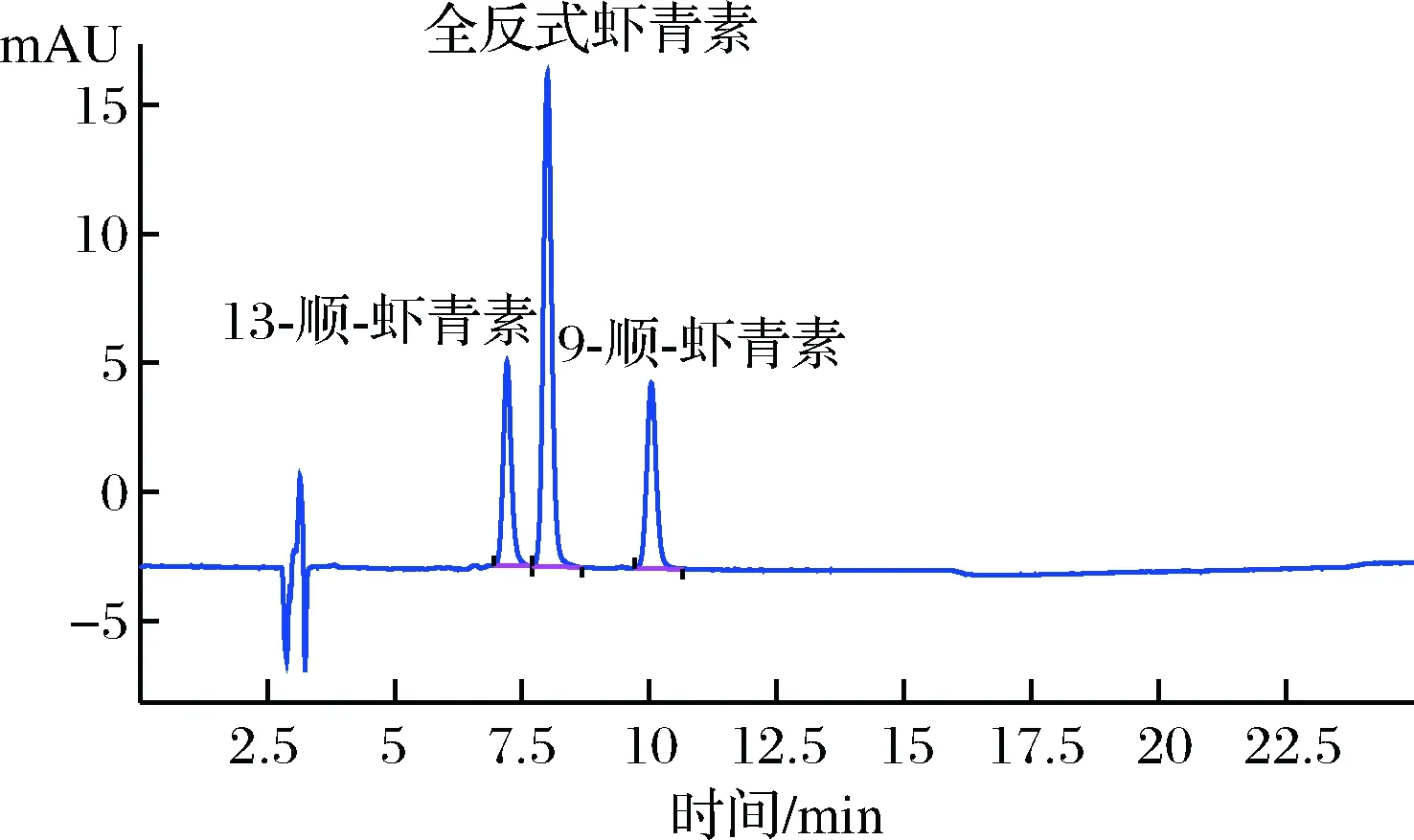
图1 全反式虾青素、9-顺式虾青素、13-顺式虾青素标准溶液液相色谱图Fig.1 Chromatograms of all trans astaxanthin,9-cis-astaxanthin,13-cis-astaxanthin
2.2 提取溶剂的选择
室温条件下,虾青素在丙酮、二氯甲烷、二甲亚砜、乙醇及其他非极性溶剂中的溶解性较好[28]。本试验根据前人研究基础,选取5种不同提取溶剂,比较不同提取溶剂对虾青素油中虾青素的提取得率,结果显示,石油醚对虾青素的提取效果最佳,显著高于其他提取溶剂(P<0.05),结果见表2,虾青素油中虾青素异构体的HPLC色谱图见图2。

表2 不同提取溶剂的提取结果(n=3)Table 2 Results of different extraction solvents(n=3)

图2 虾青素油脂中全反式虾青素、9-顺式虾青素、13-顺式虾青素液相色谱图Fig.2 Chromatograms of all trans astaxanthin,9-cis-astaxanthin,13-cis-astaxanthin in astaxanthin oil
2.3 酶解时间的选择
胆固醇酯酶的酶解时间影响虾青素酯的酶解效果,酶解时间不足,会导致酶解不完全,酶解时间过长,会造成虾青素的降解,不同酶解时间提取结果见表3。
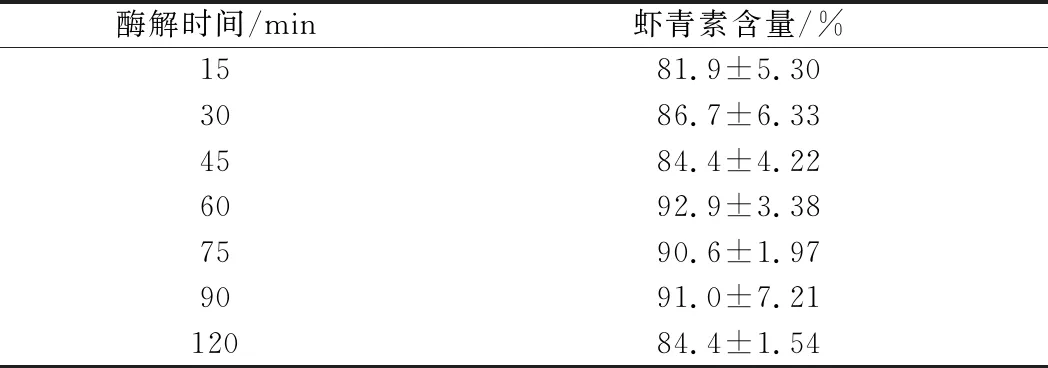
表3 不同酶解时间的提取结果(n=3)Table 1 Results of different enzymolysis time(n=3)
由表3知,在37 ℃条件下,随着酶解时间的延长,虾青素的含量增加, 60 min时酶解完全,虾青素含量为92.9%,继续酶解到75 min和90 min,虾青素含量分别为90.6%和91.0%,与60 min时没有显著性差异(P>0.05),酶解120 min,虾青素含量明显降低(P<0.05),其原因可能是温度对虾青素的降解破坏作用导致,因此酶解时间需要严格控制,以避免虾青素的损失,以4 U/mL 胆固醇酯酶37 ℃条件下酶解60 min最佳。
2.4 方法验证
2.4.1 线性关系、检出限及定量限
按照1.3.3操作,在2.1色谱条件下进行HPLC分析,得到全反式虾青素为0.10~4.00 μg/mL,浓度与峰面积有良好的线性关系,虾青素的检出限为0.1 mg/g,定量限为0.3 mg/g。
2.4.2 方法精密度实验
按实验优化的前处理方法和色谱条件,平行测定6次,虾青素油中虾青素含量的相对标准偏差(relative standard deviation,RSD)为1.36%,表明精密度高,适合虾青素的定量分析,结果见表4。

表4 方法精密度结果(n=6)Table 4 Results of precision(n=6)
2.4.3 方法准确度实验
准确度为分析方法测定的平均值与真值相符的程度。稳定样品中加入不同水平已知量的标准物质(将标准物质的量作为真值)称加标样品;同时测定虾青素油和虾青素油加标样品;加标样品扣除样品值后与标准物质的误差即为该方法的准确度,按公式(2)计算。
(2)
式中:P,加入的标准物质的回收率;m,加入的标准物质的质量,μg;X1,加标试样的测定值,μg;X0,未加标试样的测定值,μg。采用样品加标试验进行本方法准确度验证,结果见表5。
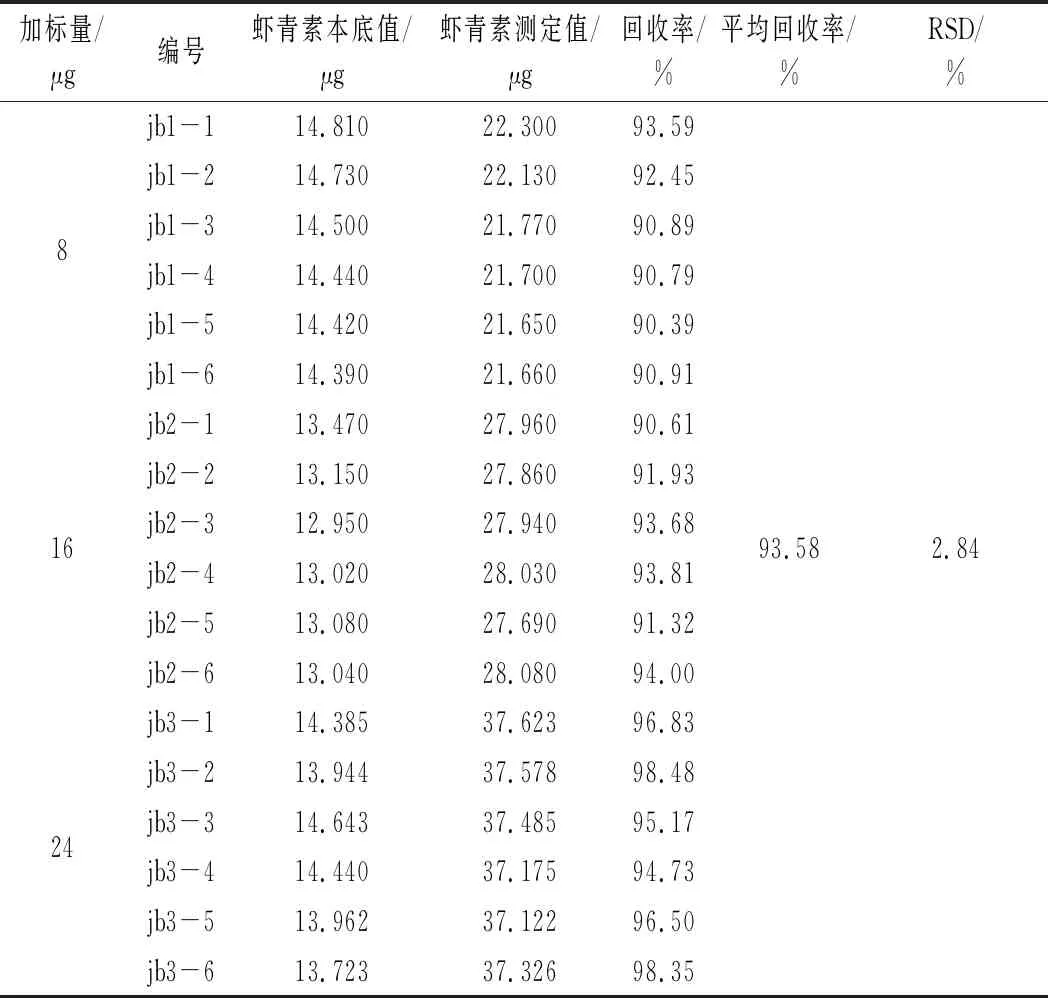
表5 加标回收率实验结果(n=6)Table 5 Results of recovery rate of ginger(n=6)
由表5可知,虾青素油中虾青素的回收率为90.39%~98.48%,平均回收率为93.58%,RSD为2.84%,表明方法添加回收率高,相对标准偏差小,适合虾青素含量的测定。
2.5 不同脱脂方法测得虾青素的含量
比较实验建立的胆固醇酯酶酶解方法、碱性皂化方法对虾青素油中虾青素的提取效率,结果见表6。

表6 不同脱脂方法测定结果(n=3)Table 6 Results of different degreasing methods(n=3)
由表6可知,胆固醇酯酶酶解所得虾青素含量是92.9%,显著高于苗凤萍[29]报道的72.04%和ZHAO等[12]报道的63.2%,与苏芳[26]报道的92.95%结果相近。酶解所得虾青素含量是碱性皂化得率的2.11倍,这可能是因为碱不仅对脂肪酸有破坏作用,对虾青素也有很大的降解作用,而胆固醇酯酶对脂肪酸有特异性,可以减少对虾青素的降解。张丽瑶等[30]研究表明虾青素溶液经超声处理后,全反式异构体的浓度大幅度降低,9-顺式和13-顺式异构体浓度均有所增加,超声处理使虾青素降解为其他成分。对比虾青素的得率,可见,酶解法对虾青素的含量影响较小,先酶解再进行高效液相色谱分析是对含虾青素酯样品的含量及几何异构体测定的可靠方法,可以准确定量样品中虾青素的含量。
3 结论
本实验在前人研究的基础上,设计单因素实验,对提取试剂、胆固醇酯酶酶解时间进行优化,以虾青素得率高低作为评价指标,得到虾青素油中虾青素的最佳提取条件为37 ℃恒温水浴振荡酶解60 min,石油醚提取,该方法对虾青素油中虾青素的提取得率为92.9%,是碱性皂化得率的2.11倍。通过比较色谱柱、流动相等高效液相色谱条件,以全反式虾青素、9-顺-虾青素、13-顺-虾青素的分离度和峰形对称性作为评价指标,筛选最佳色谱条件为YMCTMC30色谱柱,甲醇-甲基叔丁基醚两相梯度洗脱,可以实现3种虾青素异构体的有效分离,相比于目前常用的三相洗脱程序和正相洗脱系统,要求的液相系统简单。选择虾青素油进行方法学验证,结果表明,全反式虾青素在0.10~4.00 μg/mL的浓度范围内与峰面积有良好的线性关系;虾青素的检出限为0.1 mg/g,定量限为0.3 mg/g;按实验优化的最佳酶解方式和色谱条件,平行测定6次,虾青素油中虾青素含量的RSD为1.36%,表明精密度高;按虾青素油样品中虾青素含量的0.5倍、1倍、1.5倍3个浓度水平,进行3水平6平 行加标回收实验,得到虾青素油中虾青素的回收率为90.39%~98.48%,平均回收率为93.58%,RSD为2.84%,表明添加回收率高,相对标准偏差小,实验建立的方法适合虾青素油样品中虾青素含量的测定。
猜你喜欢
中南民族大学学报(自然科学版)(2022年4期)2022-07-01
轻工学报(2022年3期)2022-06-22
食品科学(2022年6期)2022-03-30
中国油脂(2022年1期)2022-02-12
中国饲料(2021年17期)2021-11-02
当代水产(2021年6期)2021-08-13
落叶果树(2021年6期)2021-02-12
中国生殖健康(2020年5期)2021-01-18
中国生殖健康(2018年5期)2018-11-06
分析化学(2017年12期)2017-12-25
