
基于5种拓扑分析方法鉴定与口腔鳞状细胞癌相关的生物标志物
2021-03-29 08:03毕文静
西北民族大学学报(自然科学版) 2021年1期
马 华,毕文静,张 旭
(西南大学 数学与统计学院,重庆 400715)
0 引言
口腔鳞状细胞癌(OSCC)是头颈部最常见的癌症.世界范围内,口腔癌约占全世界恶性肿瘤的3%[1-2].每年全世界估计有300万新病例[2-3].尽管口腔癌的治疗取得了长足进步,早期患者的生存率超过90%,但晚期患者的总体生存率仅为30%[4-5].患者不能早诊治、局部淋巴结转移及原发灶复发是口腔鳞癌预后差及五年生存率低的主要原因[2,6].因此,早期阶段癌症预测因子的检测显得十分重要.
与微阵列方法相比,转录本的测序(RNA-Seq)数据具有多个优势,包括检测体细胞突变和准确测量等位基因特异性表达能力等[7].口腔癌中最常见的细胞来源是鳞状细胞,在口腔癌中占的比例超过90%[8].晚期阶段,这些癌细胞具有高度浸润性和转移性.Wang等人通过分析来自GEO和癌症基因组图谱(TCGA)数据,发现CXCL10、OAS2、IFIT1、CCL5、LRRK2和PLAU六个基因可作为口腔鳞状细胞癌的生物标记或潜在治疗靶标[9].Wang等人通过转录组测序和生物信息学分析发现,TGFBI过表达可促进口腔鳞状细胞癌,并与患者不良预后有关[10].Ge等人通过加权基因共表达网络分析确定了七个hub基因PLAU、SERPINE1、LAMC2、ITGA5、TGFBI、FSCN1和HLF与口腔鳞状细胞癌的发生和预后相关[11].Liu等人研究表明,唾液LDOC1是口腔鳞状细胞癌的性别差异生物标志物[12].然而,上述文献中均使用微阵列芯片数据集以及采用单一的方法选择hub基因,并且OSCC的潜在分子机制尚不清楚,有待进一步探索.
本研究利用公共数据库转录本的测序(RNA-Seq)数据集,使用edgeR鉴定了口腔鳞状细胞癌组织和正常组织之间的差异表达基因,并通过STRING V11数据库构建了差异基因的PPI网络.使用Cytoscape软件对其进行可视化,采用五种拓扑分析方法确定了hub基因.CytoHubba提取了网络中与hub基因相关的重要模块.GO功能和KEGG途径富集分析探索这些聚类模块在OSCC发病机理中的作用.Kaplan-Meier分析评估了这些hub基因的预后价值.
1 材料和方法
1.1 材料
收集一组来自口腔鳞状细胞癌和它配对的正常组织的表达数据.此数据来自美国国家生物信息中心(NCBI)数据库的GEO数据(http://www.ncbi.nlm.nih.gov/geo/),下载文档GSE20116_RAW.tar.该数据集包括三个肿瘤样本和三个正常样本.文献[7]运用这组数据证明了肿瘤中的等位基因失衡与拷贝数突变相关,而拷贝数突变又与转录本丰度的变化强烈相关.本研究主要通过5种拓扑分析方法取交集筛选与口腔鳞状细胞癌相关的hub基因作为潜在的生物标志物.
1.2 方法
1.2.1 DEGs的鉴定和富集分析
使用edgeR软件包,鉴定口腔鳞状细胞癌组织和正常组织之间的DEGs.首先,采用TMM算法对数据进行标准化和过滤(相同基因符号的不同RefSeq转录本,为每个基因符号保留一个转录本).随后采用负二项分布对差异表达分析进行检验.对于任意样本i中的任意一个基因g,假设它符合二项式分布
Ygi=NB(MiPgi,φg)
(1)
其中Mi是样本i中读段总数;φg是基因g的离散度;Pgi是基因g在某个条件或分组j下的相对丰度.
筛选差异基因的标准是:将阈值设置为log2(FC)>1和p-value<0.05表示上调的差异基因,log2(FC)<-1和p-value<0.05表示下调的差异基因.使用Cytoscape中的插件Bingo来查看所有DEGs在生物过程(biological processes,BP),分子功能(molecular functions,MF)和细胞成分(cell components,CC)的中的富集情况[13].
1.2.2 PPI网络与OSCC相关聚类模块的构建
首先,把筛选出的所有DEGs输入到STRING V11数据库构建PPI网络.基于PPI网络中的基因,通过CytoHubba插件中的5种拓扑分析方法取交集选出hub基因[14].然后,利用CytoHubba插件构建与hub基因相关的聚类模块.本文将其作为OSCC相关模块.
1.2.3 GO功能和KEGG途径富集分析
为了探索OSCC相关模块中基因的潜在功能,使用R中的clusterProfiler程序包,enrichGO(pvalueCutoff = 0.01,qvalueCutoff = 0.01)和enrichKEGG(pvalueCutoff = 0.01,qvalueCutoff = 0.01)函数对这个模块执行GO功能分析和KEGG途径富集分析.ClusterProfiler是Bioconductor的R包,可以对基因集或基因簇的功能性聚类进行统计分析和可视化[15].
1.2.4 生存分析
Kaplan-Meier绘图仪数据库(http://kmplot.com) 是一种在线工具,可用于评估乳腺癌、卵巢癌、肺癌和胃癌等患者中基因的预后价值.该工具用于分析已鉴定的hub基因与总体生存率之间的关联[16].计算风险比(HR)及其0.95 的置信区间.根据基因表达量的中位值将数据分为高表达组和低表达组.使用默认参数,计算每个基因高表达组和低表达组的总体生存率.若P<0.05,则说明该基因与总体生存相关.
2 结果
2.1 DEGs的鉴定和富集分析
从GSE20116数据集中共鉴定了1 269个DEGs,包括331个上调和938个下调基因.这些差异基因的分布情况如图1.灰色表示在OSCC中上调或下调的DEGs.Cytoscape的插件Bingo生成一个有向无环图,图2颜色深浅代表富集程度,越深富集水平越高,反之越低.
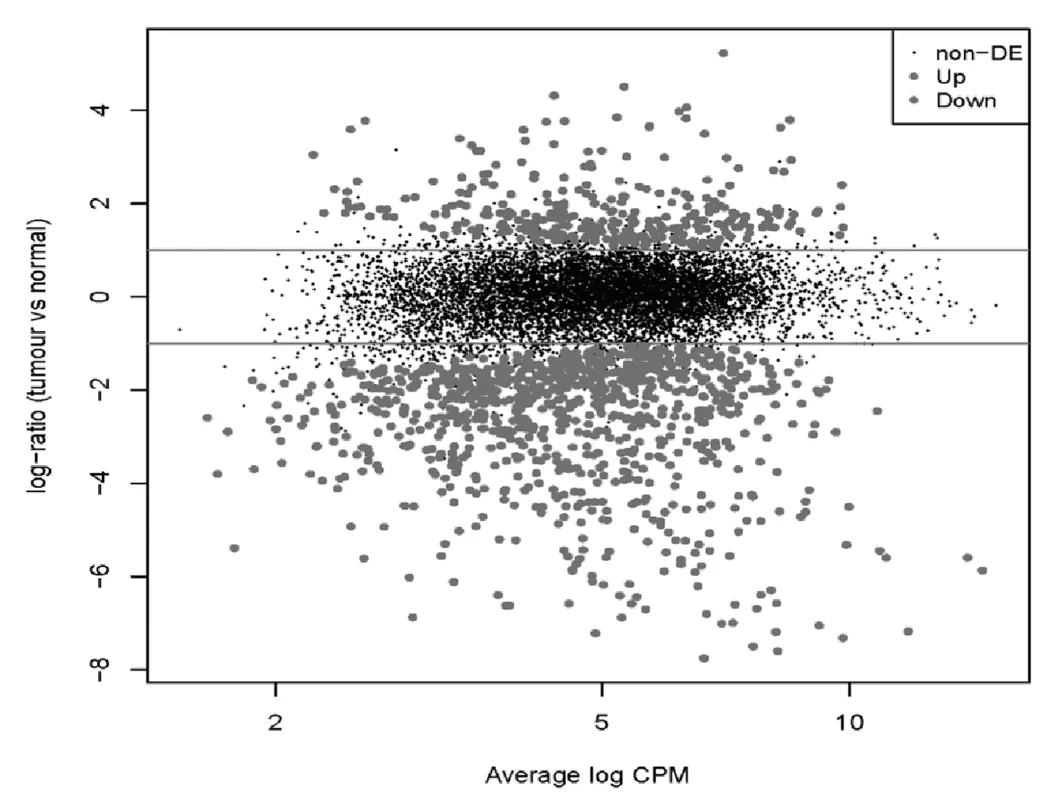
图1 差异表达基因的火山图

图2 差异表达基因的富集分析
2.2 PPI网络和OSCC相关聚类模块的构建
基于1 269个DEGs,构建了具有1 194个基因节点和7 900条边的PPI网络(图3A),其中浅灰色代表hub基因,深灰色代表差异基因.通过5种常用的计算hub基因的算法,较为准确地找到hub gene.表1是每种方法前20个重要基因的排名,然后取交集(图3B).其中被五种算法公认的hub基因是EGF、FGF2、IGF1、ACTN2、ACTA1、VWF、PTPRC、KDR、CXCL12、PTGS2和TLR4.CytoHubba插件提取了与11个hub基因相关的重要聚类模块.该模块具有393个节点和3 848条边(图3C),其中浅灰色代表hub基因,深灰色代表差异基因.把这个OSCC相关的模块用于下游途径分析.

表1 5种拓扑分析方法排名前20的基因
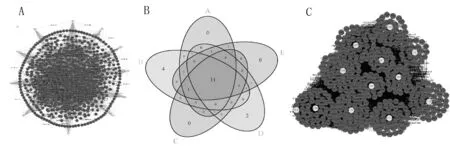
A:PPI网络;B:5种方法取交集的韦恩图;C:由11个hub基因调控的聚类模块
2.3 GO功能和KEGG途径富集分析
为了探讨这个聚类模块在OSCC发病机制中的作用,对OSCC相关模块进行了GO功能分析和KEGG通路分析(图4).其中图4A是BP中排名前15的术语、4B是CC中排名前15的术语、4C是MF中排名前15的术语和4D是KEGG途径分析中排名前15的术语.GO功能分析结果表明,855个生物过程,显著地富集在肌肉细胞分化、细胞外结构组织、细胞外基质组织、肌肉组织发育等过程;70个细胞组分主要富集在收缩纤维、肌原纤维、肌动蛋白细胞骨架、肌节细胞外基质等和52个分子功能,其中与肌动蛋白结合、肌动蛋白丝结合、细胞黏附分子结合、糖胺聚糖结合、硫化合物结合、受体配体活性等功能显著相关.而KEGG途径富集分析鉴定了35个KEGG途径,其中与PI3K-Akt信号通路、ECM-受体相互作用、黏着斑和轴突传导等显著相关.PI3K-Akt信号通路抑制剂LY2442可逆转PAR-2引起的致癌作用.PAR-2可通过激活PI3K-Akt信号通路促进OSCC的生长和发展[17-19].ECM-受体相互作用和黏着斑通路等有密切的相互作用.这些相互作用有利于OSCC的增殖、运动、分化和ECM代谢,同时抑制细胞死亡、平稳地极化生长和ECM的稳定性[20].ECM受体相互作用和黏着斑信号通路对癌细胞有明显的调控作用[21].

图4 GO功能和KEGG途径富集分析
2.4 生存分析
使用Kaplan-Meier绘图仪数据库,绘制了11个hub基因的预后价值(图5).根据基因表达量的中位值将数据分为高表达组和低表达组.灰色线条代表高表达组,黑色线条代表低表达组.log-rankP<0.05,说明高低表达组的生存率有显著差异.由图5可以看出,EGF、FGF2、IGF1、ACTN2、VWF、PTPRC、KDR、CXCL12、PTGS2和TLR4高表达组的生存率显著高于低表达组,说明这10个基因的低表达组伴有较差的总体生存率.而ACTA1低表达组的生存率显著高于高表达组,说明该基因高表达组的生存率较差.综上所述,这11个hub基因都与OSCC患者的总体生存率相关(log-rankP<0.05).

图5 hub基因的生存分析图
3 讨论
分子遗传学和信号通路的研究可以促进对OSCC发病机理的了解,并有助于OSCC的早期诊断.因此,鉴定OSCC的差异表达基因可能有助于早期诊断和开发有效的治疗方法.
在本研究中,从OSCC数据集中总共识别出1 269个DEGs,其中331个上调和938个下调基因.基于这些DEGs构建了一个PPI网络.该网络由1 194个基因和7 900个边组成.然后,通过5种拓扑分析方法取交集选择了11个hub基因.CytoHubba提取了与11个hub基因相关的聚类模块.为了探索该聚类模块在OSCC发病机理中的作用,对该模块进行了GO功能和KEGG途径分析.发现该模块中的基因富含多种功能和途径,其中PI3K-Akt信号通路、ECM-受体相互作用、细胞黏附分子(CAMs)和人乳头瘤病毒感染等与OSCC显著相关.Kaplan-Meier分析显示11个hub基因与OSCC患者的整体生存有关.
这项研究的总体目标是确定可以作为OSCC诊断和治疗的潜在生物标记的hub基因,并通过综合分析进一步探索OSCC的潜在机制.在文献研究的基础上,从生物学角度揭示了11个hub基因都与癌症相关.其中相关文献已经证实了有5个hub基因在OSCC中具有重要作用,6个hub基因与其他癌症和疾病密切相关.Chen等人研究表明,miR-23a-3p可能通过靶向FGF2抑制肿瘤的增殖、侵袭并促进OSCC的凋亡[22].Zhao等人研究表明,随着启动子甲基化率的升高,ACTN2,ACTA1在OSCC中的表达降低[23].Tand等人研究表明,VWF被用作内皮细胞标记,这种蛋白质是由内皮细胞合成的,在止血中起着重要作用,促进了第一批血小板与血管内皮下细胞的黏附[24].Zhang等人发现,CXCL12与FoxP3 +肿瘤浸润淋巴细胞相关,并影响OSCC患者的生存[25].Ikebe等人发现,TLR4增强了胰腺癌细胞的侵袭能力并促进了胰腺癌的进程[26].Li等人研究表明了miR-26b与PTGS2相互作用,抑制PTGS2蛋白表达.此外,PTGS2表达抑制细胞增殖[27].Takahashi等人研究表明,血管内皮生长因子及其受体KDR的表达与人结肠癌的血管形成、转移和增殖相关[28].Kong等人研究表明,表面活性物质(EPR)降低OSCC表皮生长因子(EGF)受体表达[29].Jacobsen等人研究表明,PTPRC的突变与某些家系中多发性硬化症的发展有关[30].Cheng等人研究表明,IGF1遗传变异与前列腺癌的发病风险相关联[31].
4 结论
针对以往筛选hub基因方法单一的问题,提出一种可以综合考虑各种方法来精确筛选hub基因的新思路.本文主要通过5种拓扑分析方法,确定了11个hub基因,包括EGF、FGF2、IGF1、ACTN2、ACTA1、VWF、PTPRC、KDR、CXCL12、PTGS2和TLR4.通过途径分析、相关文献和kaplan-meier分析验证了这些hub基因.结果表明,这11个hub基因可以作为OSCC的潜在预后生物标志物和治疗靶标.尽管对本研究结果需要进一步的生物学验证,但本研究为探索潜在的生物标志物和OSCC相关的诊断、预后和治疗靶标提供了新的思路.
猜你喜欢
祝您健康·文摘版(2020年7期)2020-07-13
心电与循环(2020年1期)2020-02-27
中国生殖健康(2019年2期)2019-08-23
世界知识(2018年19期)2018-11-21
老年教育(老年大学)(2018年12期)2018-01-28
江苏农业科学(2017年5期)2017-04-15
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
听力学及言语疾病杂志(2015年5期)2015-12-24
现代检验医学杂志(2015年5期)2015-02-06
湖北农业科学(2014年3期)2014-07-21
