
有机分子吸附和衬底调控锗烯的电子结构*
2021-03-26 08:43肖美霞冷浩宋海洋王磊姚婷珍何成
物理学报 2021年6期
肖美霞 冷浩 宋海洋 王磊 姚婷珍 何成
1) (西安石油大学材料科学与工程学院, 西安 710065)
2) (西安交通大学, 金属材料强度国家重点实验室, 西安 710049)
1 引 言
锗烯是由锗原子组成的具有类似石墨烯结构的二维材料, 与组成石墨烯的sp2杂化的碳原子不同, 锗原子在能量上更倾向于sp3杂化[1].2014 年,在Pt(111)金属表面通过超高真空表面物理气相可控生长锗原子法首次制备出单原子层锗烯[2], 随后在Au(111)[3], Al(111)[4], Cu(111)[5]等表面上也成功制备了锗烯.锗烯具有诸多优异的性质和潜在的应用, 如高的载流子迁移率、量子自旋霍尔效应和优异的抗拉伸特性等, 这使得锗烯在半导体电子器件、传感器等方面具有重要的应用潜力[6].特别是锗烯易于在现有的硅基和锗基电子工业中集成,这也是促使人们广泛关注锗烯的另一个重要因素.由于场效应晶体管中的开关电流比在很大程度上取决于沟道材料的带隙, 然而, 锗烯的微小带隙特点[7]造成场效应晶体管无法有效地关闭[8], 因此,要实现锗烯在逻辑电路中应用, 必须在锗烯材料中打开一个合适的带隙.对于场效应晶体管的室温运行, 需要高达10—100 的开关电流比, 相对应的带隙应大于100 meV[9].
目前, 表面共价功能化[10-12]、缺陷[13-15]等方法可打开类石墨烯材料的带隙, 但是损伤了载流子迁移率.对于高性能的锗烯基场效应晶体管来说,目前需要解决的问题是在不降低电子性质的前提下打开较大的带隙.最近, 研究表明表面物理吸附有机分子[16,17]可以打开类石墨烯材料的带隙并保持载流子迁移率.Wang 等[17]研究了几种有机小分子吸附对锗烯电学性质的影响, 研究发现由于有机分子破坏了锗烯亚晶格的对称性, 有效地打开了锗烯的带隙(3.9—81.9 meV), 并且保持了较小的有效质量和较高的载流子迁移率的狄拉克锥特性.然而, 对于场效应晶体管的室温运行, 只有几十meV 的带隙是远远不够的.
二维半导体纳米材料的带隙也可以通过施加外电场来进行调节.已有研究表明, FGaNH 纳米薄膜(GaN 表面镓原子进行氟化而氮原子进行氢化)的带隙在正电场作用下会显著地拓宽, 而在负电场下则迅速减小[18].双层石墨烯带隙随着外加电场强度(0—0.3 V/Å)的增强而增大, 可连续调谐到250 meV[19].锗烯和硅烯的带隙均伴随着外加垂直电场强度(0—1.03 V/Å)的增强呈现出线性增大趋势, 带隙大小可以分别达到0.012 eV 和0.016 eV[20].锗烯表面吸附甲烷和氨气后形成的甲烷/锗烯体系和氨气/锗烯体系分别在外电场强度为0—0.7 V/Å和0—0.6 V/Å的范围内可以实现大范围的线性可调谐带隙(0—69.39 meV 和37.66—134.17 meV), 更重要的是, 带隙的大小只随着外电场强度发生变化, 而与电场方向几乎无关, 而且临界电场会导致带隙重新打开和关闭[21].
此外, 二维半导体纳米材料的能带结构可以通过异质结构来进行调控.Zhang 等[22]研究发现GaAs/Ge/GaAs 量子阱由于As-Ge 界面的电荷转移不同于Ge-Ga 界面的电荷转移, 产生了强电场,由此不仅降低了Ge 的带隙, 而且诱导强烈的自旋轨道相互作用, 发生拓扑绝缘体转变.对于锗烯/GaAs 体系, 锗烯可在As-中断和Ga-中断的GaAs(111)表面稳定存在, 并呈现蜂窝状六角几何构型,然而锗烯与GaAs 衬底间存在共价键作用, 破坏了锗烯的狄拉克锥电子性质, 而利用氢插层可恢复锗烯狄拉克电子性质[23].近年来, 大量的实验和理论研究都表明, 弱相互作用不仅可以形成相对稳定的二维异质结构, 而且可以有效调节材料的电子性质.例如锗烯的带隙可进一步通过其与衬底的相互作用来调节, 同时保留狄拉克锥特性[6,24-26].然而,衬底通常只对锗烯底层配对, 因此打开带隙的程度受到限制.如果有机分子沉积在上表面, 层间可以转移更多的电荷, 由此, 界面偶极子可以进一步增强, 这可能打开更大的带隙.Gao 等[27]研究发现有机分子物理吸附以及硅烷衬底共同作用可有效拓宽硅烯的带隙, 且载流子迁移率也得到了很好的保留.Zhou 和Zhao[28]研究发现四硫富瓦烯(TTF)/锗烯/MoS2带隙在正电场作用下呈现近似线性递增趋势, 而带隙在负电场作用下呈现近似线性递减趋势.这些研究为调整锗烯的电子特性提供了有效的设计思路.
综合以上因素的考虑, 本文采用第一性原理方法研究有机分子和衬底在电场作用下调控锗烯电子性质的影响规律.首先调查了有机分子(苯和六氟苯)吸附的锗烯体系在电场作用下的原子结构以及电学性质; 接下来选择锗烷(HGeH)作为衬底,研究了有机分子吸附和衬底对锗烯电学性质的耦合作用; 最后探讨了外电场作用下有机分子/锗烯/衬底体系的电学性质变化趋势.
2 计算方法
采用基于密度泛函理论DMoL3模块[29,30]研究外电场作用下有机分子吸附和衬底对锗烯原子结构和电学性质的影响.相关交换函数用广义梯度近似中的Perdew-Burke-Ernzerhof (PBE)方法[31].核处理方法使用的是全电子效应[32], 基本设置使用双数字极化[33].因标准的PBE 泛函不能很好地描述层间或原子间弱相互作用, 本文采用DFT-D(D 表示色散)方法来研究有机分子和衬底与锗烯之间的相互作用[34], 此方法已广泛地用于存在范德瓦耳斯力作用的异质薄膜[35-39]及表面吸附小分子的纳米薄膜体系[40-42]的理论研究中.锗烯采用2 × 2 的超晶胞, 在Z 轴方向设置15 Å的真空层来避免相邻晶胞之间的相互作用, 所有原子均完全弛豫.在结构优化与性质计算中, K 点设置为17 ×17 × 1, 能量收敛公差为1.0 × 10—5Ha (1 Ha =27.2114 eV), 最大力收敛公差为0.002 Ha/Å, 最大位移收敛公差为0.005 Å.
3 结果与讨论
3.1 有机分子吸附的锗烯体系
选择吸附的有机分子为苯(C6H6)和六氟苯(C6F6).由于锗烯原子结构特点, 它通常有四种典型的吸附位置, 即空心位、Ge—Ge 键中心处、锗烯中上层Ge 原子的上方和下层Ge 原子的上方.此外, 考虑到有机分子取向及其高对称几何结构, 本文从理论上针对在锗烯表面吸附有机分子形成的苯/锗烯体系和六氟苯/锗烯体系构建了八种有机分子吸附构型, 如图1 所示.T-1 和T-3 构型的有机分子/锗烯体系中C—C 键均垂直于Ge—Ge 键,T-2 和T-4 构型的有机分子/锗烯体系中C—H (F)键均平行于Ge—Ge 键, H-1 构型的有机分子/锗烯体系中C—H (F)键垂直于Ge—Ge 键, H-2 构型的有机分子/锗烯体系中C—H (F)键平行于Ge—Ge键, B-1 构型的有机分子/锗烯体系中C—C键垂直于Ge—Ge 键, B-2 构型的有机分子/锗烯体系中C—H (F)键平行于Ge—Ge 键.有机分子/锗烯体系通过吸附能计算结果来确定实验上可行的有机分子吸附构型, 并获得最稳定的原子结构.这种建模思路及方法已应用于硅烯表面吸附TTF 分子体系[27]和锗烯表面吸附四氰基苯分子(TCNB)体系[43]中.
当有机分子吸附在锗烯上时, 通过(1)式计算吸附能Ead:

其中, Ead表示锗烯表面上吸附单个有机分子的吸附能; E有机分子表示每个苯分子或六氟苯分子的能量; Ef-锗烯(s-锗烯)表示独立的锗烯(衬底支撑的锗烯)的能量; E总体表示表面吸附有机分子的锗烯体系的总能量.根据(1)式, 当吸附能Ead越大时, 表明有机分子和锗烯之间结合越强.表1 列出了苯/锗烯体系和六氟苯/锗烯体系的吸附能Ead、吸附距离H、翘曲高度d 和带隙Eg.

图1 有机分子吸附在2 × 2 锗烯超晶胞表面的八种吸附构型示意图(粉色球和蓝色球分别表示锗烯中上层锗原子和下层锗原子, 灰色球表示C 原子, 而白色球表示H 原子或F 原子)Fig.1.Schematic view of the eight adsorption configurations of the organic molecules adsorbed on the 2 × 2 supercell of germanene, where the pink balls represent the upper Ge atoms of the germanene, the blue balls represent the lower Ge atoms of the germanene, the gray balls represent the C atom, and the white balls represent the H or F atoms.
由表1 可知, 苯/锗烯体系的最稳定的构型为T-1, 而六氟苯/锗烯体系的最稳定的构型为T-4.图2(a)和图2(b)给出了T-1 构型的苯/锗烯体系和T-4 构型的六氟苯/锗烯体系的原子结构图.T-1构型的苯/锗烯体系的吸附能Ead约为0.676 eV,比其他构型体系的吸附能大了0.014—0.154 eV;T-4 构型的六氟苯/锗烯体系的吸附能Ead约为0.656 eV, 比其他构型体系的吸附能大了0.009—0.135 eV.同时, T-1 构型的苯/锗烯体系比T-4 构型的六氟苯/锗烯体系的吸附能大了0.020 eV.研究结果表明, 锗烯吸附苯和六氟苯的吸附能均比锗烯吸附乙炔(0.160 eV)、乙醇(0.406 eV)、甲醇(0.325 eV)、甲烷(0.114 eV)及氨分子(0.444 eV)[21]大, 这说明苯和六氟苯更容易吸附在锗烯表面上.
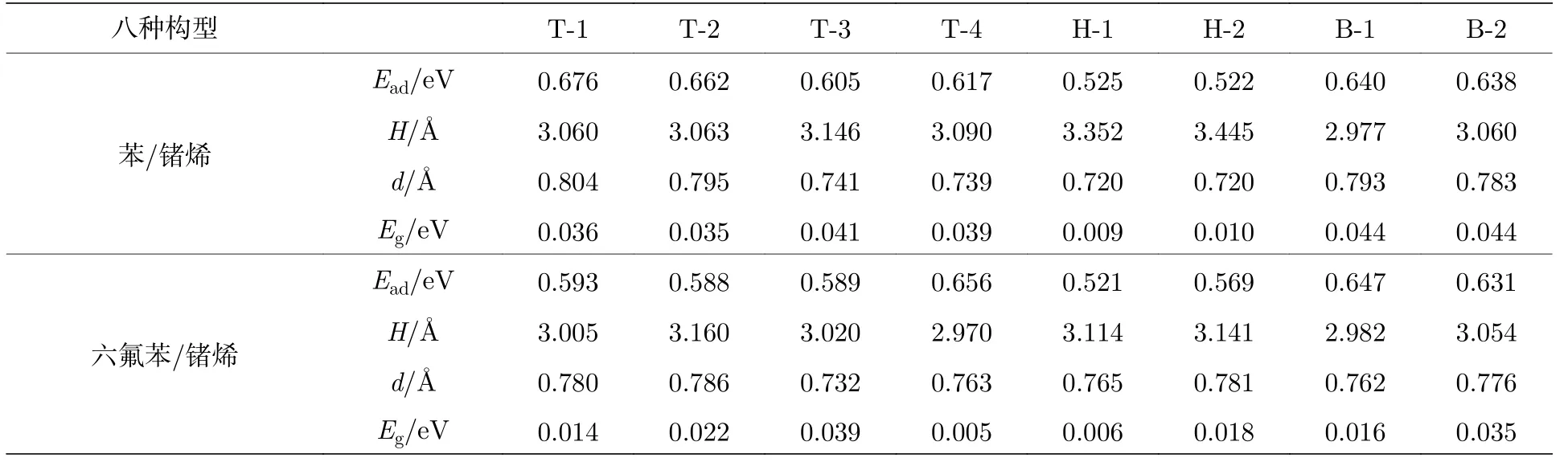
表1 苯/锗烯体系和六氟苯/锗烯体系的八种高对称吸附构型的吸附能Ead、吸附距离H、翘曲高度d 和带隙EgTable 1.Adsorption energy Ead, adsorption distance H, buckling height d and band gap Eg of eight highly symmetric adsorption configurations of benzene/germanene and hexafluobenzene/germanene systems.
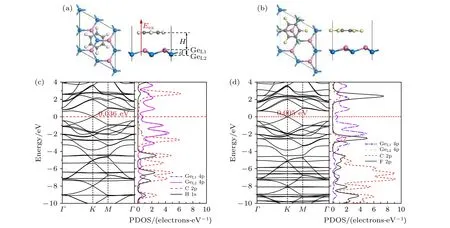
图2 (a) T-1 构型的苯/锗烯体系的俯视图和主视图.(b) T-4 构型的六氟苯/锗烯体系的俯视图和主视图.粉色球和蓝色球分别表示锗烯上层和下层Ge 原子, 灰色球表示C 原子, 白色球和黄色球分别表示H 原子和F 原子; Eex 表示垂直于锗烯的外电场强度, 从锗原子指向有机分子方向的电场为正电场, 反之为负电场.(c) T-1 构型的苯/锗烯体系的能带结构图和部分态密度(PDOS)图.(d) T-4 构型的六氟苯/锗烯体系的能带结构图和部分态密度图Fig.2.(a) Top and side views of the benzene/germanene systems with T-1 configuration.(b) The top and side views of the hexafluorobenzene/germanene systems with T-4 configuration.The pink and blue balls represent the upper and lower Ge atoms of the germanene, respectively.The gray balls represent the C atom.The white and yellow balls represent the H and F atoms, respectively.External electric field Eex perpendicular to the germanene is applied along the upward direction (defined as positive “+”, i.e.,the electric field direction is from the germanene to the organic molecules at positive electric field) or the downward direction(defined as negative “—”).(c) Band structure and partial density of states (PDOS) of benzene/germanene with T-1 configuration.(d) Band structure and PDOS of hexafluobenzene/germanene with T-4 configuration.
定义吸附距离H 为体系结构优化后有机分子与锗烯上层GeL1原子之间的最小距离.苯/锗烯体系中苯吸附距离H 为2.970—3.445 Å; 六氟苯/锗烯体系中六氟苯吸附距离H 为3.070—3.160 Å.此外, 有机分子吸附会导致锗烯发生一定的变形, T-1构型的苯/锗烯体系中锗烯翘曲高度d 为0.804 Å,说明结构变形越大, 吸附能越大, 结构也越稳定.然而, T-4 构型的六氟苯/锗烯体系虽然比其他几种构型的吸附能都大, 但锗烯的翘曲高度(d =0.763 Å)却不是最大的.与原始锗烯的翘曲高度(d = 0.694 Å)相比, 苯吸附后导致锗烯结构变形较大, 这再次说明了苯对锗烯的吸附作用比六氟苯的吸附作用强.苯/锗烯体系或六氟苯/锗烯体系的吸附能和吸附距离都表明了苯分子或六氟苯分子和锗烯之间并没有形成化学键, 只是产生弱的层间交互作用.由此, 不同的吸附构型的苯/锗烯体系和六氟苯/锗烯体系的原子结构和电子性质是相似的.本文只研究了最稳定的T-1 构型的苯/锗烯体系和T-4 构型的六氟苯/锗烯体系在外电场作用下的原子结构和电学性质.
为了研究有机分子对锗烯电学性质的影响, 计算了苯/锗烯体系以及六氟苯/锗烯体系的能带结构.独立的锗烯具有微小带隙特点[7].如图2(c)和图2(d)所示, 当锗烯表面吸附苯分子或六氟苯分子后, 狄拉克点周围的能带结构几乎不受影响,K 点附近的线性色散仍然存在, 这表明在吸附有机分子后, 载流子的迁移率可以很大程度上保持.由于有机分子和锗烯之间弱的相互作用, 可以粗略地将能带结构看成是锗烯和苯分子或六氟苯的简单结合, 并且可以观察到T-1 构型的苯/锗烯体系打开了约0.036 eV 的直接带隙, 而T-4 构型的六氟苯/锗烯体系打开了约0.005 eV 的直接带隙.其他构型的苯/锗烯体系和六氟苯/锗烯体系打开带隙的情况如表1 所列.该打开带隙现象类似于TTF/硅烯[27]中观察的现象.图2(c)和图2(d)还分别给出了T-1 构型的苯/锗烯体系和T-4 构型的六氟苯/锗烯体系的部分态密度(PDOS)图.研究结果表明, 苯/锗烯体系和六氟苯/锗烯体系的价带顶和导带底均主要由GeL14p 轨道和GeL24p 轨道决定,并且苯和六氟苯与锗烯之间存在弱的交互作用.
为了证实苯/锗烯体系和六氟苯/锗烯体系中锗烯保留了较高的载流子迁移率, 通过
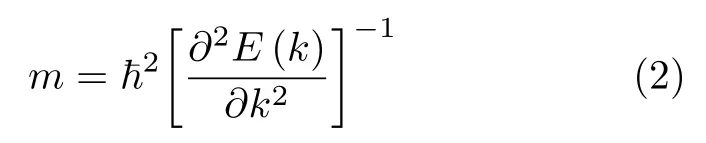
计算电子有效质量me和空穴有效质量mh, 采用

计算电子和空穴两种载流子的迁移率μe和μh.这里ħ表示普朗克常数, k 表示波矢量, E(k)表示色散关系, τ 表示散射时间.
对于原始的锗烯, μe和μh的值分别为6.24 ×105cm2·V—1·s—1和6.54 × 105cm2·V—1·s—1[44].假设散射时间τ 与原始锗烯相同.由(2)式和(3)式计算得到, 苯/锗烯体系中me和mh分别是原始锗烯的3.49 和3.41 倍, μe和μh的值分别为1.79 × 105cm2·V—1·s—1和1.92 × 105cm2·V—1·s—1; 六氟苯/锗烯体系的me和mh分别是原始锗烯的3.21 和3.13倍, μe和μh的值分别为1.94 × 105cm2·V—1·s—1和2.09 × 105cm2·V—1·s—1.研究结果表明, 表面吸附苯或六氟苯的锗烯体系保留了较高的载流子迁移率.
为了定量分析有机分子和锗烯之间的电荷转移量, 采用Mulliken 布居分析, T-1 构型的苯/锗烯体系中C6H6分子向锗烯转移了0.029e, 这表明苯分子是一个供体分子, 并且在锗烯亚晶格中上层Ge 原子失去了0.177e, 而下层Ge 原子得到了0.206e; 而T-4 构型的六氟苯/锗烯体系中C6F6分子从锗烯中得到了0.007e, 说明六氟苯分子是一种受体分子, 并且在锗烯亚晶格中上层Ge 原子失去了0.305e, 而下层Ge 原子得到了0.298e.该研究结果表明, 有机分子吸附打破了锗烯中上层Ge 原子和下层Ge 原子之间的电荷分布平衡, 从而锗烯中A 和B 亚晶格周围的电子密度分布不均匀, 最终导致两个亚晶格之间不再等效.因此, 根据紧束缚模型, K 点处的带隙可被打开[45].
对稳定的T-1 构型的苯/锗烯体系和T-4 构型的六氟苯/锗烯体系分别施加外电场Eex(电场方向如图2 所示), 研究了外电场对表面吸附有机分子的锗烯体系的电子性质的影响.图3 给出了T-1构型的苯/锗烯体系和T-4 构型的六氟苯/锗烯体系的带隙Eg随外电场强度变化的规律图.可以清楚地观察到, 这两种体系的带隙Eg均随着负电场或正电场强度Eex的增大呈现出近似线性增加的趋势.值得注意的是, 表面吸附有机分子的锗烯带隙的大小主要与外电场的强弱有关, 几乎与外电场方向无关.对于T-1 构型的苯/锗烯体系, 在负电场作用下, 当Eex达到—0.90 V/Å时, 带隙值可增加到0.680 eV; 在正电场作用下, 当Eex达到0.80 V/Å时, 带隙值可达到0.593 eV.当负电场或正电场的强度再增加时, T-1 构型的苯/锗烯体系的直接带隙均将转变为间接带隙, 甚至从半导体转变为导体特性.对于T-4 构型的六氟苯/锗烯体系,在负电场作用下, 当Eex达到—1.00 V/Å时, 带隙值可增加到0.521 eV; 在正电场作用下, 当Eex达到0.80 V/Å时, 带隙值可以达到0.505 eV.随着负电场或正电场的强度增加, T-4 构型的六氟苯/锗烯体系的直接带隙也将逐渐减小, 甚至转变为间接带隙半导体, 最终转变为导体.该现象与甲烷/锗烯体系和氨气/锗烯体系分别在外电场强度为0—0.7 V/Å和0—0.6 V/Å范围时呈现线性可调谐带隙(0—69.39 meV 和37.66—134.17 meV)的变化趋势相同[21].值得注意的是, 原始锗烯的带隙Eg在外电场作用下呈现线性增加趋势[20], 当外电场强度Eex达到1.03 V/Å时, 锗烯可打开约0.12 eV的带隙, 而苯/锗烯体系和六氟苯/锗烯体系在低于此电场强度情况下可分别打开约0.680 eV 和0.521 eV 的直接带隙, 这表明在外电场作用下表面吸附苯和六氟苯有机分子可有效拓宽锗烯的带隙.
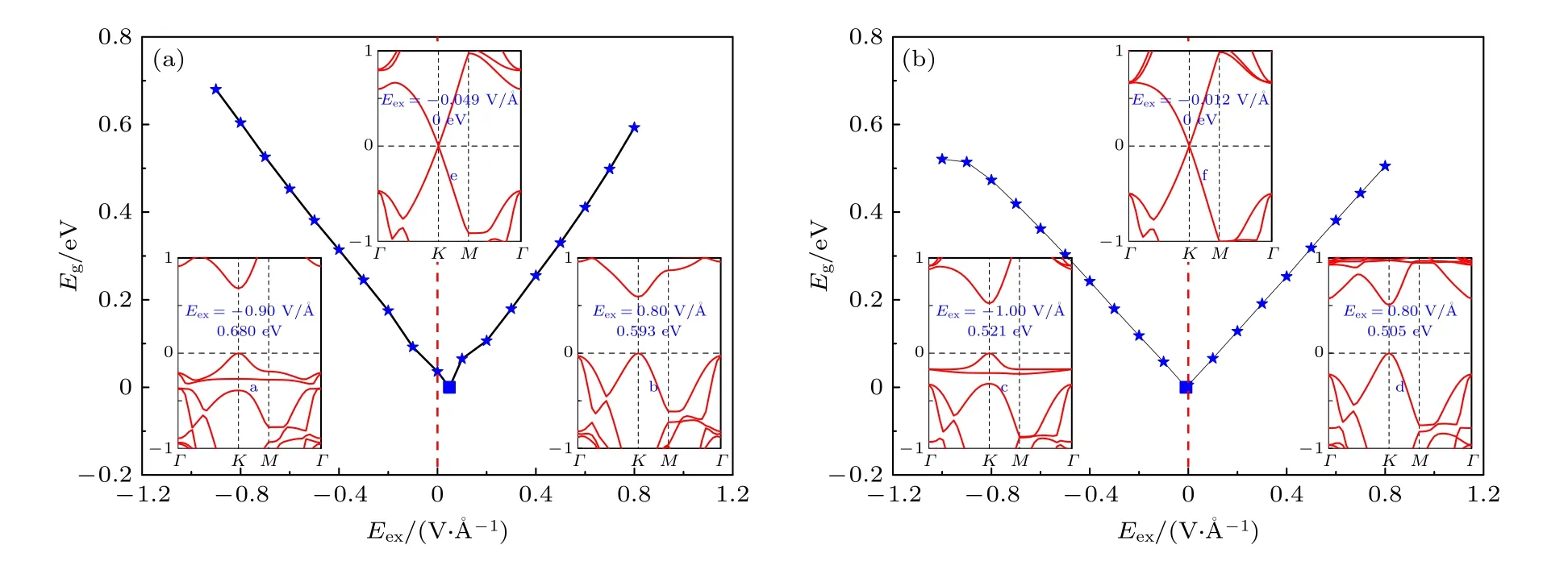
图3 (a) T-1 构型的苯/锗烯体系和(b) T-4 构型的六氟苯/锗烯体系的带隙Eg 随外电场强度Eex 的变化图, 插图a—d 分别显示了两种体系在负电场和正电场作用下具有最大带隙时的能带结构, e 和f 分别显示了两种体系在临界外电场下具有零带隙时的能带结构图Fig.3.Band gaps Eg of (a) benzene/germanene system with T-1 configuration and (b) hexafluorobenzene/germanene system with T-4 configuration as a function of the strength of the external electric field.The inserts a—d show the band structures of both systems with the maximum band gap under negative and positive electric field, while e and f show the band structure of both systems with the zero-gap under the critical external electric field.
进一步分析T-1 构型的苯/锗烯体系和T-4 构型的六氟苯/锗烯体系在外电场作用下的电荷转移情况, 如图4 所示.在负电场作用下, 苯分子和六氟苯分子为受体分子, 当外电场强度Eex达到一定值时, 锗烯中下层GeL2原子失去的电子逐渐增加,而有机分子(苯或六氟苯)以及锗烯中上层GeL1原子得到的电子逐渐增加; 在正电场作用下, 苯分子和六氟苯为供体分子, 有机分子以及锗烯中上层GeL1原子失去的电子会逐渐增加, 而锗烯中下层GeL2原子得到的电子也将逐渐增加.由此可知,尽管外电场作用导致有机分子以及锗烯内部之间电荷转移方向不同, 但是伴随着Eex的不断增强,它们之间的电荷转移量Q 都逐渐增加, 导致锗烯亚晶格之间的电荷分布不均更加显著, 从而促使T-1 构型的苯/锗烯体系和T-4 构型的六氟苯/锗烯体系在外电场作用下可打开更大的带隙.
有机分子/锗烯体系的带隙Eg和电场强度E的依赖关系可以通过K 点的双带模型[46]解释, 哈密顿量公式为
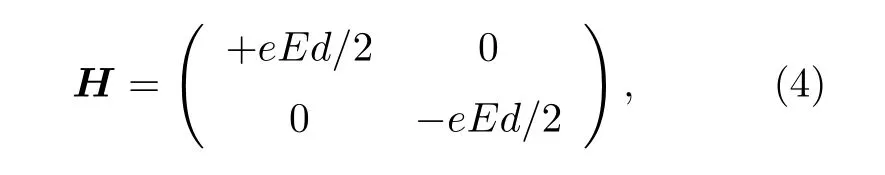
其中, E 为电场强度, d 为翘曲高度.带隙Eg可以通过公式Eg= eEd 进行计算, 当电场强度E = 0时, 带隙值为零.然而, 在外电场强度Eex为零时,苯/锗烯体系和六氟苯/锗烯体系带隙Eg分别为0.036 eV 和0.005 eV.因此, (4)式中的电场强度E应为复合电场强度, 包括内电场Ein和外电场Eex.由于表面吸附的有机分子和锗烯之间存在电荷转移, 这可以采用电容器模型来进行讨论[21].有机分子吸附诱导的内电场Ein与有机分子电荷转移量Q成 正 比, 可 以 通 过 公 式Ein= 2Q/[ε0a2sin(π/3)]计算, 其中Q 表示转移电荷量, ε0表示真空电容率, a 表示锗烯晶格常数.图5 给出了不同外电场强度Eex下T-1 构型的苯/锗烯体系和T-4 构型的六氟苯/锗烯体系内电场强度Ein.研究发现,当外电场强度Eex为零时, 苯/锗烯体系和六氟苯/锗烯体系的内电场Ein分别约为—0.046 V/Å和0.011 V/Å.为了验证此理论, 详细模拟计算了苯/锗烯体系和六氟苯/锗烯体系在外加负电场(—0.1—0 V/Å)和外加正电场(0—0.1 V/Å)下的电学性质, 如图3(a)和图3(b)所示, 发现苯/锗烯体系和六氟苯/锗烯体系分别在外加正电场(0.049 V/Å)和外加负电场(—0.012 V/Å)作用下带隙为零.

图5 (a) T-1 构型的苯/锗烯体系和(b) T-4 构型的六氟苯/锗烯体系的内电场强度Ein 与外电场强度Eex 变化关系图Fig.5.Strength of internal electric field Ein of (a) benzene/germanene system with T-1 configuration and (b) hexafluorobenzene/germanene system with T-4 configuration as a function of the strength of external electric field Eex.
如图4(a)和图4(b)所示, 在外电场作用下,伴随负电场强度的增加, 苯分子和六氟苯分子为受体分子, 电子从锗烯转移到有机分子, 内电场Ein方向由锗烯指向有机分子; 而随着正电场强度的增加, 苯分子和六氟苯分子为供体分子, 电子从有机分子转移到锗烯, 导致内电场方向从有机分子指向锗烯.如图5(a)和图5(b)所示, 尽管内电场方向始终与外电场方向相反, 并且Ein随着Eex的增加也逐渐增大, 但是在稍大外电场强度Eex作用下,Ein绝对值小于Eex绝对值, 由此复合电场强度E将与外电场方向一致, 并且随着外电场强度Eex增加而增大.而有机分子/锗烯体系中锗烯的翘曲高度d 受外电场强度Eex的影响变化不大.依据公式Eg= eEd, 本文结果清晰地解释了苯/锗烯体系和六氟苯/锗烯体系的带隙Eg随外电场强度Eex呈现近似线性增加趋势的原因.
3.2 有机分子/锗烯/锗烷体系
锗烷(GeH4)为无色、剧毒、可自燃、非腐蚀性气体; 热稳定性较差, 大约在280 ℃就能检测到GeH4分解为锗和氢, 在350 ℃下GeH4几乎全部分解成单质锗和氢气; GeH4的自催化性很强, 一旦分解形成了金属覆盖膜, 就会急剧分解, 故其分解爆炸危险性很高[47].GeH4作为高纯单质锗的重要原材料, 主要应用于电子器件及太阳能电池等领域.另一种锗烷(HGeH)为表面完全氢化的锗烯,具有类似石墨烷的结构, 是一种新型二维材料, 并且可通过表面共价功能化来调节其直接带隙[48,49].HGeH 在室温下具有18000 cm2·V—1·s—1的高迁移率、非零带隙以及稳定性等优点, 促使其在电子和光电子器件等领域, 特别是场效应晶体管方面, 具有重要的应用前景[50].
本文选择HGeH 作为衬底材料来研究有机分子和衬底对锗烯原子结构和电学性质的影响, 主要有以下两个方面的原因.一方面, HGeH 是一种类似于锗烯晶体结构的二维半导体材料, 其晶格常数为4.05 Å, 而锗烯的晶格常数为4.02 Å, 它们之间的晶格错配度只有0.75%, 比锗烯与GaAs 衬底[23]、锗烯与InSe 衬底[26]的错配度(1.30%和2.00%)更小, 这更容易形成匹配的异质结构[8].将锗烯与HGeH 衬底的结合能(Ec)定义为Ec= E锗烯/HGeH—Ef-锗烯— EHGeH, 其中E锗烯/HGeH表示锗烯/HGeH体系的总能量, Ef-锗烯表示原始锗烯的能量, EHGeH表示HGeH 衬底的能量.研究发现单元超晶胞锗烯与HGeH 衬底的结合能Ec为—0.292 eV, 这表明锗烯与HGeH 衬底之间存在一定的交互作用力,这有利于形成锗烯/HGeH 异质结构.另一方面, 研究发现锗烯与GaAs 衬底间存在共价键作用, 破坏了锗烯的狄拉克锥电子性质[23], 而锗烯和HGeH之间不存在共价键作用, 而是通过弱的相互作用形成稳定的双层异质结构.HGeH 衬底仅破坏了锗烯亚晶格的对称性, 从而促使锗烯在狄拉克点上打开了带隙[6], 并且很好地保留了锗烯的高载流子迁移率.计算得到的锗烯/HGeH 体系的载流子迁移率(1.56 × 105cm2·V—1·s—1)略大于锗烯/InSe 体系的载流子迁移率(1.42 × 105cm2·V—1·s—1)[26].由于不同堆垛模式的锗烯/HGeH 体系原子结构和电学性质相似, 本文在最稳定的锗烯/HGeH 体系原子结构基础上研究有机分子和衬底对外电场下锗烯电学性质的调控作用.

图6 (a)苯/锗烯/HGeH 体系的俯视图和主视图.(b)六氟苯/锗烯/HGeH 体系的俯视图和主视图, 其中粉色球和蓝色球表示锗烯上层和下层Ge 原子, 灰色球表示C 原子, 白色球和黄色球表示有机分子中H 原子和F 原子, 橙色球和黑色球表示HGeH 上层H 原子和下层H 原子, 绿色球和红色球表示HGeH 上层和下层Ge 原子; Eex 表示垂直于锗烯的外电场, 从HGeH 指向有机分子方向的电场为正电场, 反之为负电场.(c)苯/锗烯/HGeH 体系的能带结构图和部分态密度(PDOS)图.(d)六氟苯/锗烯/HGeH 体系的能带结构图和部分态密度图Fig.6.(a) Top and side views of the benzene/germanene/HGeH system.(b) Top and side views of the hexafluorobenzene/germanene/HGeH systems.The pink and blue balls represent the upper and lower Ge atoms of the germanene, respectively.The gray balls represent the C atom.The white and yellow balls represent the H or F atoms of organic molecules.The orange and black balls represent the upper and lower H atoms of HGeH.The green and red balls represent the upper and lower Ge atoms of HGeH,respectively.External electric field Eex perpendicular to the germanene is applied along the upward direction (defined as positive“+”, i.e., the electric field direction is from the HGeH to the organic molecules at positive electric field) or the downward direction(defined as negative “—”).(c) Band structure and PDOS of benzene/germanene/HGeH.(d) Band structure and PDOS of hexafluobenzene/germanene/HGeH.
图6 (a)和图6(b)给出了苯/锗烯/HGeH 体系和六氟苯/锗烯/HGeH 体系最稳定的原子结构, 其中有机分子苯和六氟苯最稳定的吸附位置依然分别为T-1 构型和T-4 构型(如图1 所示).根据(1)式可得出, 苯/锗烯/HGeH 体系的吸附能为 Ead=0.708 eV, 比苯/锗烯体系吸附能(0.676 eV)大了0.032 eV.相比苯/锗烯体系, 苯/锗烯/HGeH 体系中苯分子的吸附距离H 缩短到2.989 Å, 锗烯和HGeH 间的最小层间距D 为1.525 Å, 锗烯的翘曲高度d 增大到0.886 Å.六氟苯/锗烯/HGeH 体系的吸附能Ead增大为0.706 eV, 相应的吸附距离H减小到2.959 Å, 锗烯和HGeH 间的最小层间距D为1.650 Å, 锗烯的翘曲高度d 增大到0.777 Å.这一现象说明了有机分子/锗烯/HGeH 体系的吸附能越大, 锗烯的结构变形越大.
图6(c)和图6(d)分别给出了苯/锗烯/HGeH体系和六氟苯/锗烯/HGeH 体系的能带结构和部分态密度图.由于锗烯与HGeH 衬底存在交互作用, 这进一步影响了K 点周围的抛物线色散关系,苯/锗烯/HGeH 体系的直接带隙增加到0.152 eV,而六氟苯/锗烯/HGeH 体系的直接带隙增加到0.105 eV.该直接带隙的增大主要是由衬底和吸附的有机分子共同影响的.分析部分态密度图可以发现, 尽管有机分子和衬底对锗烯的电子结构有一定的影响, 但是苯/锗烯/HGeH 体系和六氟苯/锗烯/HGeH 体系中费米面附近的导带底和价带顶均主要由GeL24p 轨道和GeL14p 轨道决定.通过(2)式和(3)式计算得出苯/锗烯/HGeH 体系中载流子迁移率μe和μh的值分别为1.36 × 105cm2·V—1·s—1和1.31 × 105cm2·V—1·s—1, 而六氟苯/锗烯/HGeH体系中载流子迁移率μe和μh的值分别为1.53 ×105cm2·V—1·s—1和1.51 × 105cm2·V—1·s—1.该结果表明, 在锗烷(HGeH)衬底和有机分子共同作用下锗烯依然可以在很大程度上保持载流子的迁移率.
进一步采用Mulliken 布局分析, 对于苯/锗烯/HGeH 体系, C6H6分子失去0.035e, 锗烯中下层GeL2原子失去0.338e, 上层GeL1原子得到0.409e,HGeH 衬底失去0.036e.而对于六氟苯/锗烯/HGeH 体系, C6F6分子得到0.006e, 锗烯中下层GeL2原子失去0.209e, 上层GeL1原子得到0.242e,HGeH 衬底得到0.039e.在两种体系中, 有机分子以及HGeH 衬底导致锗烯中两个亚晶格之间电荷分布不均更加剧烈.该电荷转移现象很好地解释了有机分子/锗烯/HGeH 体系中带隙增大的原因.
对苯/锗烯/HGeH 体系和六氟苯/锗烯/HGeH体系的稳定结构施加垂直电场(电场方向如图6 所示), 研究电场对两种体系电学性质的影响.图7 给出了苯/锗烯/HGeH 体系和六氟苯/锗烯/HGeH体系带隙Eg随外加电场强度Eex的变化趋势.研究结果表明, 在负电场作用下, 苯/锗烯/HGeH 体系的直接带隙Eg随着外电场强度Eex的增加而增加, 当Eex为—0.35 V/Å时, 直接带隙Eg达到最大值(0.463 eV), 而外加负电场强度继续增大时, 该体系最终会转变为导体.而对于六氟苯/锗烯/HGeH体系, 在负电场作用下, 其直接带隙Eg将会继续增加, 当Eex为—0.35 V/Å时, 直接带隙Eg达到最大值(0.358 eV), 而外加负电场强度进一步增大时, 该体系将转变为导体.值得注意的是, 苯/锗烯/HGeH 体系和六氟苯/锗烯/HGeH 体系均在较小外加正电场强度(0.1 V/Å)下从直接带隙半导体转变为导体.该现象说明了有机分子/锗烯/HGeH体系带隙不仅与外电场强度有关, 也与外电场方向有关.

图7 (a)苯/锗烯/HGeH 体系和(b)六氟苯/锗烯/HGeH 体系的带隙Eg 随外电场强度Eex 的变化关系图, 其中插图显示了两个体系具有最大带隙时的能带结构Fig.7.Band gaps Eg of (a) benzene/germanene/HGeH system and (b) hexafluorobenzene/germanene/HGeH system as a function of the strength of the external electric field Eex.The inserts show the band structures of both systems with the maximum band gaps.

图8 (a)苯/锗烯/HGeH 体系和(b)六氟苯/锗烯/HGeH 体系的电荷转移量Q 随外电场强度Eex 变化关系图Fig.8.Charge transfer Q of (a) benzene/germanene/HGeH system and (b) hexafluorobenzene/germanene/HGeH system as a function of the strength of external electric field Eex.
图8 给出了苯/锗烯/HGeH 体系和六氟苯/锗烯/HGeH 体系在外电场作用下的电荷转移情况.研究结果表明, 在外电场作用下, 两种体系均表现出从HGeH 到锗烯的电荷转移现象.对于外加负电场作用下苯/锗烯/HGeH 体系, 尽管苯分子在外电场强度Eex为—0.2—0 V/Å时为供体分子, 电子从苯分子转移给锗烯/HGeH 体系, 可能抵消苯/锗烯/HGeH 的界面偶极子, 但是HGeH 转移给锗烯的电荷量远远大于苯与锗烯之间的电荷转移量, 诱发外加负电场作用下锗烯中上层GeL1原子得到电子和下层GeL2原子失去电子的电荷量均单调线性增加, 这加剧了锗烯亚晶格间的电荷分布不均现象, 进一步增大了苯/锗烯/HGeH 体系的带隙Eg.对于外加负电场作用下六氟苯/锗烯/HGeH 体系,六氟苯分子始终为受体分子, 它从锗烯中获取电子, 这进一步增加了界面偶极子, 并且随着负电场强度的增加, 锗烯中上层GeL1原子和下层GeL2原子间的电荷分布不均加剧, 最终促使六氟苯/锗烯/HGeH 体系的带隙Eg进一步拓宽.值得注意的是,在相同的负电场强度下, 苯/锗烯/HGeH 体系中锗烯亚晶格间电荷分布不均比六氟苯/锗烯/HGeH体系的情况更为强烈, 导致苯/锗烯/HGeH 体系可打开相对较大的带隙Eg.该现象实际上是HGeH衬底、表面吸附的有机分子和电场的协同效应.相反, 在外加正电场作用下, 两种体系中有机分子和HGeH 均向锗烯转移电子, 削弱了有机分子/锗烯/HGeH 的界面偶极子, 从而使体系带隙Eg随正电场强度Eex的增加而减小, 最终苯/锗烯/HGeH 体系和六氟苯/锗烯/HGeH 体系均在0.1 V/Å下关闭带隙.
4 结 论
本文采用基于密度泛函理论并考虑范德瓦耳斯修正的第一原理模拟方法, 研究了有机分子吸附和衬底对电场作用下锗烯原子结构和电子性质的影响.研究结果表明, 由于有机分子吸附和衬底通过弱的层间交互作用打破了锗烯亚晶格的对称性,苯/锗烯体系可打开0.036 eV 的带隙, 而六氟苯/锗烯体系打开0.005 eV 的带隙; 在有机分子和锗烷(HGeH)衬底共同作用下, 苯/锗烯/HGeH 体系带隙增大到0.152 eV, 六氟苯/锗烯/HGeH 体系带隙增大到0.105 eV; 载流子的迁移率可以很大程度上保持.在一定的外电场强度作用下, 由于有机分子和HGeH 衬底的电荷转移量增加以及体系内电场等因素导致有机分子/锗烯体系以及有机分子/锗烯/HGeH 体系的带隙可伴随外电场强度的增加呈现近似线性增大趋势.这些研究结果为调控锗烯电子性质提供了有效的设计方法, 为锗烯在场效应管和其他纳米电子学器件中的应用提供有力的理论支持.
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
成都信息工程大学学报(2019年3期)2019-09-25
科技创新与应用(2018年21期)2018-09-14
中国宝玉石(2018年3期)2018-07-09
振动工程学报(2017年4期)2018-05-31
电子制作(2018年1期)2018-04-04
北京航空航天大学学报(2017年10期)2017-04-20
中国科技信息(2016年6期)2016-08-31
中国科技信息(2015年24期)2015-11-07
