淫羊藿素制备方法、制剂新技术研究进展
2021-03-25 11:16孙彦君陈豪杰韩瑞杰郝志友冯卫生
中成药 2021年3期
孙彦君 ,陈豪杰,赵 晨,韩瑞杰,陈 辉,郝志友,冯卫生*
(1.河南中医药大学呼吸疾病中医药防治省部共建协同创新中心,河南 郑州 450046;2.河南中医药大学药学院,河南 郑州 450046;3.河南省中药特色炮制技术工程研究中心,河南 郑州 450046)
淫羊藿为小檗科植物淫羊藿Epimedium brevicornu、箭叶淫羊藿Epimedium sagittatum、柔毛淫羊藿Epimedium pubescens或朝鲜淫羊藿Epimedium koreanum的干燥叶,在我国最早记载于《神农本草经》,已有二千多年的药用历史,具有补肾阳、强筋骨、祛风湿功效[1],临床上常用于治疗骨质疏松、绝经后综合征、乳房肿块、高血压和冠心病[1-2],近年来还在抗肿瘤、抗衰老、调节骨代谢、提高免疫力、治疗艾滋病等方面显示了独特的疗效[2]。淫羊藿素是从淫羊藿中分离得到的代表性异戊烯基化黄酮醇类化合物,具有广泛药理作用,如抗肿瘤[3]、抗骨质疏松和骨骼肌萎缩[4-5]、抗阿尔茨海默病[6]、抗氧化[7]、抗抑郁[8]、调节免疫[9]、抗炎[9]、延缓肝纤维化[10]等,并尚未发现该成分对正常细胞和组织有明显的药物毒性[2,10],故其开发前景广泛,作为治疗晚期肝细胞癌的药物已进入Ⅲ期临床研究。本文对近年来关于淫羊藿素制备方法、制剂新技术的文献进行综述,以期为该成分的大量制备及合理开发利用提供参考。
1 理化性质
淫羊藿素别名阿可拉定、淫羊藿苷元,分子式C21H20O6,相对分子质量368.126 0,黄色针状结晶(甲醇),熔点为228~230 ℃,不溶于水,易溶于DMSO、乙醇、甲醇、乙酸乙酯等有机溶剂,FeCl3、Mg-HCl 反应呈阳性。EI-MSm/z(%):368[M]+(100)、353 (80)、325 (10)、312(50)。UV (MeOH) λmaxnm:216sh、250、270、304、368。1H-NMR (400 MHz,DMSO-d6)δ:12.38 (1H,s,5-OH),10.76 (1H,s,7-OH),9.49 (1H,s,3-OH),6.30(1H,s,H-6),7.13 (2H,d,J=9.0 Hz,H-3′,5′),8.13(2H,d,J=9.0 Hz,H-2′,6′),3.44 (2H,d,J=6.7 Hz,H-1″),5.18 (1H,t,J=6.7 Hz,H-2″),1.75 (3H,s,H-4″),1.63 (3H,s,H-5″),3.85 (3H,s,3-OCH3);13CNMR (100 MHz,DMSO-d6)δ:146.6 (C-2),136.4 (C-3),176.7 (C-4),158.8 (C-5),98.3 (C-6),161.7 (C-7),103.5 (C-8),153.9 (C-9),106.1 (C-10),124.1 (C-1′),129.6 (C-2′),114.6 (C-3′),160.9 (C-4′),114.6 (C-5′),129.6 (C-6′),25.9 (C-1″),122.9 (C-2″),131.5(C-3″),21.7 (C-4″),18.3 (C-5″),55.9 (3-OCH3)[11-13]。
2 提取分离
淫羊藿素主要存在于小檗科植物中,也可见于大戟科、十字花科植物,具体见表1。
淫羊藿素由于在药材中的含量极低,故一般要采用多种色谱技术分离才能保证其纯度,制备方法复杂,成本较高,很难应用于工业化生产。大花淫羊藿全草粉末(1.2 kg) 采用甲醇提取,提取物经反复ODS、Sephadex LH-20、硅胶等柱色谱分离后,得到淫羊藿素(18.3 mg)[14]。淫羊藿粉末(20 kg) 采用95%乙醇提取,回收乙醇后加水混悬,加入乙酸乙酯萃取,萃取部位经硅胶柱色谱、制备HPLC 分离,得到淫羊藿素(11 mg)[15]。巫山淫羊藿地上部位粉末(2.3 kg) 用95%乙醇提取,提取物依次用二氯甲烷、乙酸乙酯、正丁醇萃取,其中乙酸乙酯部位经聚酰胺柱色谱分离,得到淫羊藿素(36 mg)[11]。箭叶淫羊藿叶粉末(4.0 kg) 用水回流提取,浓缩后提取液经HPD 大孔吸附树脂、硅胶、Sephadex LH-20 等柱色谱结合制备HPLC 分离,得到淫羊藿素(75.3 mg)[16]。E.elatum的根和地上部位粉末(1.0 kg) 用乙醇提取,提取物经硅胶柱色谱、制备HPLC分离,得到淫羊藿素(11.4 mg)[17]。粗毛淫羊藿地上部位粉末(2 kg) 采用95%乙醇提取,回收溶剂后经氯仿、乙酸乙酯、正丁醇萃取,乙酸乙酯、正丁醇萃取部位经硅胶柱色谱分离,得到淫羊藿素(30 mg)[18]。M.siamensis枝和叶粉末(3.3 kg) 用二氯甲烷提取,提取物经硅胶柱色谱、Sephadex LH-20 柱色谱、制备型TLC 分离,得到淫羊藿素(100 mg)[19]。尾叶血桐枝粉末(10 kg) 用90%乙醇提取,提取物经乙酸乙酯萃取,萃取物经MCI 柱色谱、硅胶柱色谱、制备HPLC 色谱分离,得到淫羊藿素 (3.4 mg)[20]。荠菜地上部位粉末(11 kg) 用70%乙醇提取,提取物依次用石油醚、乙酸乙酯、正丁醇萃取,乙酸乙酯萃取部位经硅胶柱色谱、中压液相色谱、Sephadx LH-20 柱色谱分离,得到淫羊藿素(12.25 mg)[21]。
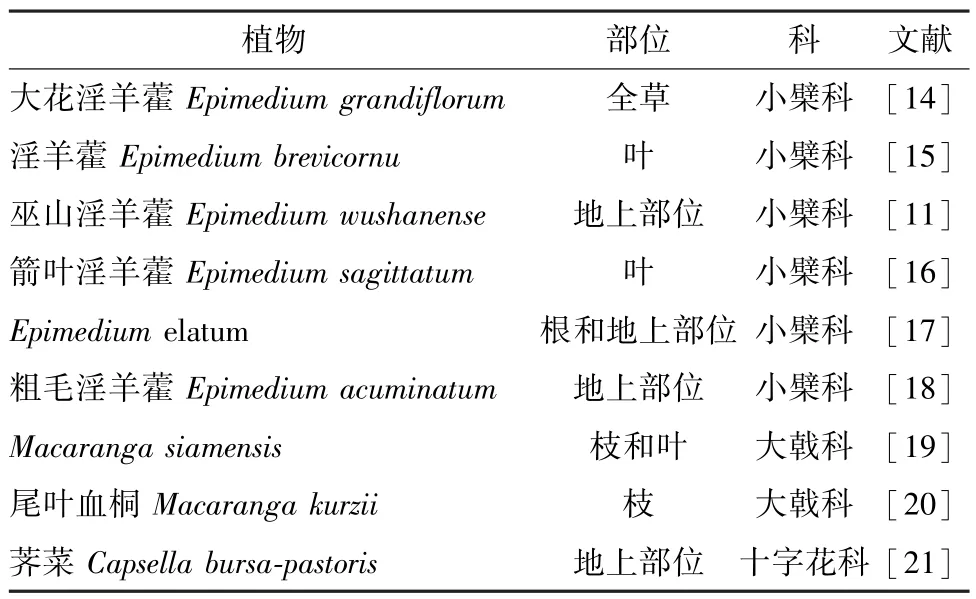
表1 淫羊藿素植物分布
3 淫羊藿苷或淫羊藿总提取物的水解
与直接从淫羊藿中提取分离相比,水解后再提取分离淫羊藿素时产量高,成本低。淫羊藿含有大量以淫羊藿苷为主的异戊烯基化黄酮苷类化合物,淫羊藿苷结构中含有2 个糖基,分别为7 位的葡萄糖基、3 位的鼠李糖基,经去糖基化处理可得到淫羊藿素。从淫羊藿中制备淫羊藿素的思路主要有2 种,一种是先将总黄酮提取出来再水解,水解产物经进一步分离得到;另一种是先将淫羊藿苷提取分离出来,再水解得到。目前,通过水解得到淫羊藿素的方法主要有酶水解、酸水解、酸-酶混合法。
3.1 酶水解 酶水解法条件温和,专属性强,所用到的酶主要有β-葡萄糖苷酶、α-L-鼠李糖苷酶、柚苷酶、蜗牛酶、果胶酶、纤维素酶等。孟坤[22]发明了β-葡萄糖苷酶制备淫羊藿素的方法,即采用水煎煮法提取淫羊藿,浓缩后的提取液经D101 大孔吸附树脂,依次用水、30%乙醇、50%乙醇洗脱,收集50%乙醇洗脱液,得到总黄酮提取物,再采用硅胶柱色谱分离,三氯甲烷-甲醇梯度洗脱,收集含有淫羊藿苷的流份,甲醇重结晶纯化,得到淫羊藿苷,通过β-葡萄糖苷酶酶解脱去糖基,得到淫羊藿素。许明淑等[23]发明了柚苷酶酶解法,即在淫羊藿苷中加入30%乙醇,再加入1 mol/L NaOH 溶液调pH 至4.0,加入柚苷酶,在50 ℃下搅拌30 h,得到淫羊藿素。贾晓斌等[24]发明了蜗牛酶酶解法,即将淫羊藿用30%~95%乙醇或30%~100%甲醇回流提取,回收溶剂,膏状物中加入pH 3.5~9.0 的醋酸-醋酸钠缓冲液和蜗牛酶,在15~80 ℃下反应2~72 h,得到淫羊藿素,重结晶得到纯品。孟坤[25]发明了果胶酶酶解法,即将淫羊藿提取物在果胶酶的作用下进行酶解反应,得到淫羊藿素。董登祥[26]发明了纤维素酶酶解法,即将淫羊藿苷溶于醋酸-醋酸钠缓冲溶液(pH=4.5),在37 ℃下搅拌15 min,加入纤维素酶酶解过夜,得到淫羊藿素。萧伟等[27]发明了一种能降解淫羊藿苷母核结构上鼠李糖残基的α-L-鼠李糖苷酶和葡萄糖残基的β-葡萄糖苷酶,通过糖苷酶组合物将淫羊藿苷转变为淫羊藿素。赵林果等[28]采用α-L-鼠李糖苷酶、β-葡萄糖苷酶组成的双酶体系,转化淫羊藿总黄酮(淫羊藿苷、朝藿定A、朝藿定B、朝藿定C)中的多组分黄酮苷而生成淫羊藿素。
3.2 酸水解董登祥[26]将淫羊藿苷在酸性条件下(5 mol/L盐酸、5 mol/L 硫酸、80%冰醋酸、体积比3 ∶10的5 mol/L 硫酸与80%冰醋酸混合液) 于60 ℃水浴中回流24 h,水解液冷却后加入NaHCO3溶液调pH 至6,乙醚萃取,回收溶剂,得到淫羊藿素。孟坤等[29]用乙醇回流提取淫羊藿,提取液经HPD-300 大孔吸附树脂纯化,用水、20%乙醇洗脱除去杂质后,加70%乙醇洗脱,收集洗脱液,得到含淫羊藿苷的浓缩物,加入四氢呋喃-异丙醇溶液、浓硫酸,在55~62 ℃下水解24 h 后加入5%NaHCO3溶液淬灭,回收有机溶剂,反应液离心,得到沉淀,加入丙酮,在50 ℃下搅拌溶解,滤过,滤液加水升温到64 ℃至溶液澄清,降温至10 ℃保温2 h,收集沉淀,得到淫羊藿素。
3.3 酸解与酶解混合法 参考文献[30] 报道。淫羊藿苷先加入5%稀硫酸在50 ℃下水解24 h,得到脱去鼠李糖基的产物淫羊藿次苷I,加入β-葡聚酶,在50 ℃下于0.1 mol/L醋酸/醋酸钠缓冲液中酶解20 h,得到淫羊藿素。
淫羊藿苷的酶解反应可选用纤维素酶或葡聚糖酶,加入纤维素酶,在37 ℃下于0.1 mol/L 醋酸/醋酸钠缓冲液(pH=5) 中水解5~6 d,生成去掉葡萄糖基的淫羊藿次苷Ⅱ、部分淫羊藿次苷Ⅰ的混合物,再加入β-葡聚糖酶,在50 ℃下于0.1 mol/L 醋酸/醋酸钠缓冲液(pH=5) 中水解20 h,水解淫羊藿次苷Ⅱ,水解产物加入5% 稀硫酸在50 ℃下水解24 h,得到淫羊藿素。
通过反复试验证实,淫羊藿苷结构中的7 位葡萄糖苷键易被酶水解,而3 位鼠李糖苷键易被酸水解。因此,该成分先经β-葡聚糖酶水解后再用5%硫酸水解,不仅制备方法简便,而且收率较高。
4 全合成与结构修饰
牟关敏等[31]以间苯三酚为原料,合成了淫羊藿素。具体合成过程(图1) 为间苯三酚与乙腈经Houben-Hoesch 反应,得到乙酰化的间苯三酚(1),再经甲氧甲基醚保护之后得到化合物2,后者与茴香醛在氢氧化钠、双氧水的催化下通过一锅法直接构建了黄酮醇母体,即5,7-二-O-(甲氧基甲基) -4′-甲氧基黄酮醇(3),脱除5-位保护基、引入3-位甲氧基甲基后得到化合物5,在碳酸钾、冠醚催化下与异戊烯基溴反应,生成5-O-异戊烯基化产物(6),加入氯苯、催化剂三-(6,6,7,7,8,8,8-七氟-2,2-二甲基-3,5-辛二酮) 铕,在80 ℃下反应,得到8 位异戊烯基化的化合物7。在盐酸作用下脱除保护基,得到淫羊藿素,总收率为4.2%。在整个合成路线中,黄酮醇骨架构建和异戊烯基重排是影响总收率的关键步骤,而这两步反应产率的提高则是淫羊藿素全合成路线优化的突破点。
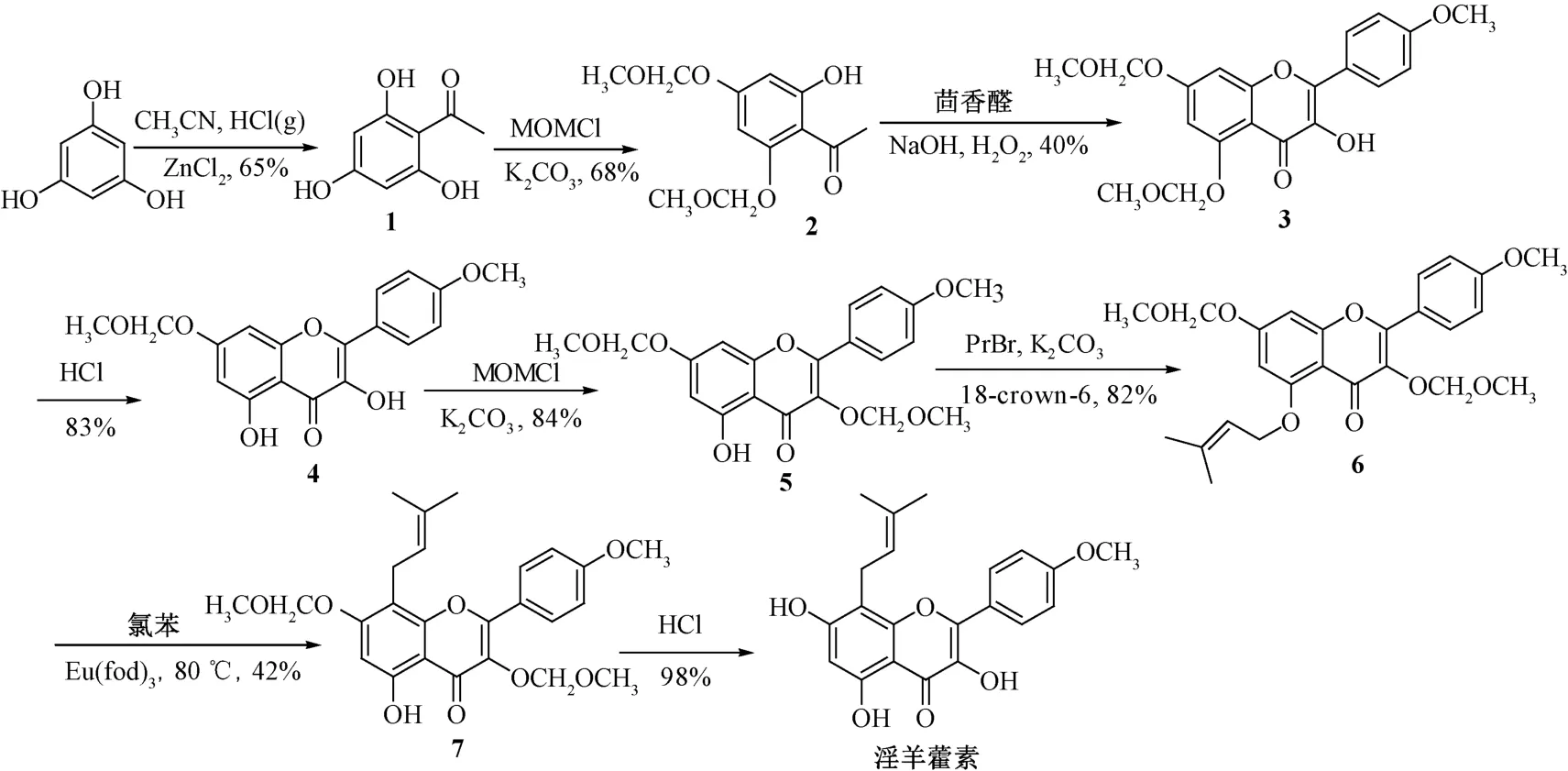
图1 淫羊藿素合成路线(Ⅰ)
Nguyen 等[12]以2,4,6-三羟基苯乙酮和对羟基苯甲酸为原料,合成了淫羊藿素。具体合成过程(图2) 为2,4,6-三羟基苯乙酮在K2CO3催化下,与BrBn 反应生成化合物1,对羟基苯甲酸与硫酸二甲酯先在NaOH 催化下后在SOCl2催化下生成4-甲氧基苯甲酰氯(2),化合物1、2 经贝克-文卡塔拉曼(Baker-Venkataraman) 重排反应和脱水闭合构建黄酮骨架,得到化合物3,与二甲基二环氧乙烷(DMDO) 反应24 h 后再与对甲苯磺酸反应2 h,得到黄酮醇骨架,即化合物4,在Pd/C 催化下脱去苄基后再进行甲氧甲醚化,得到化合物6,在K2CO3催化下与3,3-二甲基丙烯溴反应,生成5-O-异戊烯基化产物(7),在N,N-二乙基苯胺催化下得到8 位异戊烯基取代的衍生物(9),在盐酸催化下脱去保护基,得到淫羊藿素。
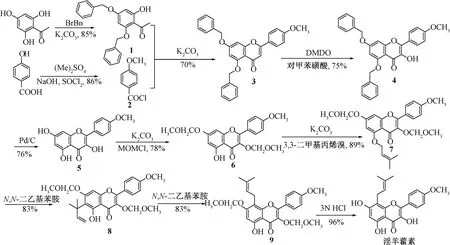
图2 淫羊藿素合成路线(Ⅱ)
Mei 等[32]以山柰酚为先导化合物进行结构修饰,得到淫羊藿素。具体合成过程(图3) 为将山柰酚中的所有羟基乙酰化,得到产物(1),与苄基溴反应生成7 位苄醚衍生物(2),在碱性条件下与硫酸二甲酯反应,得到4′位醚化同时脱去3、5 位乙酰基的产物(3),在N,N-二异丙基乙胺的催化下3 位羟基被甲氧甲醚化,得到产物(4),在碳酸钾、冠醚的催化下,与异戊烯基溴反应,生成5-O-异戊烯基化产物(5),加入氯苯、催化剂三-(6,6,7,7,8,8,8-七氟-2,2-二甲基-3,5-辛二酮) 铕,在85 ℃下反应,得到8 位异戊烯基化的化合物6,在盐酸作用下脱除3位保护基,在1,4-环己二烯、Pb/C 催化化下脱去7 位苄基,得到淫羊藿素。

图3 淫羊藿素合成路线(Ⅲ)
5 制剂新技术
淫羊藿素制成软胶囊后供三期临床使用,具有保存期长、稳定性高、生产成本低、适用于工业化生产等优点[33],但该成分水溶性、脂溶性均很差,在水中的平衡溶解度仅约为10 μg/mL[34],在大豆油、C6-C12 脂肪酸甘油酯中的溶解度也分别只有2.858、1.840 mg/mL[35]。另外,淫羊藿素以常规口服方式给药后,由于肝脏首过消除效应(主要代谢产物为葡萄糖醛酸衍生物[36]) 会降低该成分治疗指数,同时胃肠道功能和其内容物也会影响其疗效的稳定性,导致体内吸收差,生物利用度低(仅4.33%[36]),制约了其临床应用。为提高淫羊藿素溶解性、稳定性、靶向性,增强其药效,药学工作者转向了相关新剂型的研制开发。
脂质体传递系统具有良好的生物相容性,以及毒性小、安全性高、具有缓释性和靶向性等诸多优势,经口服进入胃肠道后,可将包埋的水溶性或脂溶性有效成分扩散到小肠上皮黏膜细胞,提高人体对有效成分的吸收能力。为了解决淫羊藿素在水相体系里溶解性差、受外界环境影响易发生降解等问题,并提高该成分在胃肠道消化过程中的稳定性,袁芳等[37]采用薄膜分散结合冰浴超声或者高压均质乳化处理,制备脂质体口服制剂;贾晓斌等[38]发明了一种淫羊藿素脂质体的制备方法,由大豆卵磷脂(50%~85%)、胆固醇(10%~35%)、淫羊藿素(2%~25%) 组成,表面为磷脂双分子层,双分子层内部、外部为亲水基团,双分子层之间为疏水基团,淫羊藿素包埋在脂质体的磷脂双层中,可促进该成分吸收,并且大豆卵磷脂来源广泛,成本低,具有良好的生物相容性,可生物降解。
利用纳米化技术将淫羊藿素包裹于脂质体的磷脂双分子层中时,可提高了该成分水溶性,同时该剂型物理稳定性良好,放置较长时间后未见分层、絮凝,并可选择性地聚集于单核吞噬细胞系统,给药后肝脏中药物浓度增加,能显著降低对其他脏器和组织的毒性及用量。脂质体粉针是固体制剂,可解决普通脂质体聚集、沉降、融合、渗漏等的不稳定性问题,李君[39]等发明了具有肝脏靶向性的淫羊藿素脂质体粉针,由淫羊藿素 (0.01%~5%)、磷脂(0.05%~8%)、增塑材料(0~8%)、支持剂(0~20%)组成。固体脂质纳米粒具有和脂质体一样良好的缓控释作用和生物相容性,同时可减小了聚合物纳米材料所带来的潜在毒性,黄汉[40]等研制的淫羊藿素固体脂质纳米粒包封率较高,可增强其水溶性和稳定性,提高生物利用度。
固体分散体是将药物以分子、胶态、微晶或无定形态分散在某一固态载体物质中形成的分散体,从而改善其溶解度。吴玲[34]将淫羊藿素与载体泊洛沙姆188 混匀,加热至熔融并搅拌均匀,冷却,固化,脆化,得到固体分散体,可显著提高了该成分溶出性能,而且操作简单,成本低。
磷脂具有一定的亲水性和亲脂性,药物与其形成复合物后能改善前者溶解性,增加在胃肠道中的吸收,提高生物利用度,从而提高疗效。贾晓斌等[41]将淫羊藿素磷脂复合物制成颗粒剂、片剂、胶囊剂、口服液、注射剂、丸剂、冻干粉针剂、软胶囊等剂型;孟坤等[35]将淫羊藿素与磷脂复合物制成乳液注射剂,可增加该成分脂溶性(与原料药相比,磷脂复合物在油中的溶解度提高了2 倍左右) 和化学稳定性,升高血药浓度。
孟坤等[33]将淫羊藿素制成口服制剂,即为混悬剂的软胶囊。王新峦等[42]发明了一种以淫羊藿素为活性成分的生物复合材料,用药过程中该成分微球或纳米粒从支架材料中持续释放,可长期在骨缺损局部保持疗效,从而促进骨生长。
6 结语
淫羊藿素药理作用广泛,活性高于其他淫羊藿苷元,并且毒副作用小,发展潜力大,但迄今除了抗肿瘤外,对该成分其他活性的研究仍局限于细胞、动物模型阶段,其制备方法及新剂型开发已成为影响后续开发的关键因素。由于淫羊藿素在药材中含量低,故通过提取、色谱分离得到,但其操作复杂,成本高;关于该成分合成的文献报道也较少[12,31-32],并且步骤长,总收率低,其中黄酮醇骨架的构建、异戊烯基的引入至为关键;关于水解淫羊藿苷来获取该成分的文献报道较多,主要包括酸水解、酶水解。
淫羊藿素水溶性、脂溶性均较差,如何应用当代制剂新技术改善该成分口服生物利用度,使其转变为更高效、更强效的制剂已成为药剂学领域的研究热点。随着人们对淫羊藿素药理作用机制、制备方法、新剂型开发的不断深入探索,该成分有望在抗癌、防癌、防治骨质疏松、神经保护等领域得到应用,从而造福于人类。
猜你喜欢
江西中医药(2022年8期)2022-08-22
环球中医药(2021年3期)2021-04-07
中成药(2018年12期)2018-12-29
天然产物研究与开发(2018年10期)2018-11-06
天然产物研究与开发(2018年10期)2018-11-06
中国中医药现代远程教育(2018年22期)2018-02-09
湖南林业科技(2017年6期)2018-01-30
中成药(2017年4期)2017-05-17
中国医学科学院学报(2015年5期)2015-03-01
天然产物研究与开发(2014年6期)2014-04-27