
BioMnOx对非甾体药物氧化协同Fe(III)吸附的研究
2021-03-22 06:37秦松岩吕务娟赵立新
哈尔滨工业大学学报 2021年5期
秦松岩,吕务娟,黄 馨,胡 杰,罗 义,赵立新
(1.天津理工大学 环境科学与安全工程学院,天津 300384;2.南京大学 环境学院,污染控制与资源化研究国家重点实验室,南京 210093)
乙酰氨基酚(acetaminophen,APAP)具有解热镇痛的作用,是全世界使用量最高的非甾体抗炎药物[1],通过生物代谢物及过期或未用完废弃物进入环境,导致其在污水处理厂、自然水环境、土壤中频频检出,APAP在污水处理厂的出水中质量浓度可达到8 025 ng/L[2].APAP是中国近45个污水处理厂活性污泥的主要污染物之一[3].含有微量的APAP水体经过加氯消毒后会产生毒性更大的苯醌类物质[4];APAP在水环境药物风险评价中风险值大于1,质量浓度超出了无生态风险浓度值[5-6],其残留已对水生生态系统和人体构成潜在危害[7-9],对其去除成为环境研究中的一个热点问题[10].目前,国内外研究用于降解APAP的方法主要有高级氧化法、TiO2光催化、生物降解[11-16]等.其中,生物降解法由于不存在二次污染以及成本低等备受关注.
生物锰氧化物是由锰氧化细菌如恶臭假单胞球菌(PseudomonasPutida)、纤发菌属(Leptothrix)、锰土微菌(Pedomicrobiummanganicum)等以O2为电子受体,通过多铜氧化酶(MCOs) 将Mn(II)氧化为Mn(IV)而形成高价态锰生物氧化物(BioMnOx).BioMnOx作为锰氧化细菌形成的初级产物,基本单元是 Mn(Ⅳ)O6的正八面体结构,存在包括Mn(Ⅳ)空缺位点和晶体的边缘及内层位点,Mn(IV)/Mn(II)间的氧化还原电位及强氧化剂Mn(III)为中间体的单电子转移,使其具有较强的氧化能力[17].因此,BioMnOx同时展现了较强的氧化和吸附能力.Pseudomonassp.作为锰氧化的模式菌株,其形成的锰氧化物已应用到多种重金属和有机物氧化去除的研究中.Owen[18]研究了PseudomonasputidaGB-1产生的BioMnOx对草铵铁的吸附,发现Fe(III)在多个位点特异性地吸附到矿物结构上.菅之舆等[19]在以锰氧化细菌Pseudomonassp.QJX-1构建的生物体系中,通过Pseudomonassp.QJX-1生成的BioMnOx有效去除2-羟基-4-甲氧基二苯甲酮-5-磺酸(BP-4).Zhang等[7]通过在锰砂/石英砂好氧生物滤池启动时引入Pseudomonassp.形成BioMnOx生物膜,对卡马西平、双氯芬酸等通过开苯环降解.以往研究基于对实际工艺的模拟,对Pseudomonas的不同菌株BioMnOx在水处理反应体系中,或对有机物氧化、或对重金属吸附进行了宏观评价.然而,实际水体中存在着多种金属离子,在被吸附占据吸附位点同时对BioMnOx氧化有机物的影响鲜有研究.另外,BioMnOx具有较高的吸附及氧化活性,其微观结构和活性会随时间快速变化,Pseudomonas的菌株活性、BioMnOx的形成过程及结构变化等对吸附和有机物的降解影响相关性研究,对于生物铁锰反应器的参数设计及启动运行具有重要意义.
本研究以具有强锰氧化能力的Pseudomonasputida作为模式菌种, 以水体中常见的Fe(III)为共存离子,通过研究P.putida形成锰氧化物的过程对Fe(III)的吸附及对APAP降解效率,确定BioMnOx吸附过程对氧化能力的影响;提出BioMnOx生物量对APAP降解的动力学并推断APAP的降解途径,为BioMnOx应用于水体残留非甾体药物类的去除提供新思路及理论支撑.
1 实 验
1.1 实验试剂
实验中所用药品有乙酰氨基酚(acetaminophen,APAP)(纯度>99.0%,上海润捷化学试剂有限公司)、四氢呋喃(99.9%,J&K Chemical)、N-O双(三甲基硅烷基)三氟乙酰胺(BSTFA,99%,TCI),其他无机试剂均为分析纯或优级纯,甲醇、乙腈等有机试剂均为色谱纯.
1.2 生物锰氧化物的形成
实验所用菌株为恶臭假单胞菌(PseudomonasputidaQYS-1,CGMCCNO:14390),从某除铁锰的生物滤池中筛选分离出来.
培养基成分(g/L):蛋白胨 0.8,酵母浸粉 0.2,K2HPO40.1,MgSO4·7H2O 0.2,NaNO30.2,CaCl20.1,NH4Cl 0.1,MnSO4·H2O 0.2.配置一定量的培养基, 然后用饱和 NaOH 调节 pH(7.1~7.2),分装后放于灭菌器中 121 ℃灭菌 30 min. 然后将其转入生物安全柜中, 冷却至室温.取100 mL装入锥形瓶中,将PseudomonasputidaQYS-1母液以1%的接种量接入,然后将其置于恒温振荡器中培养(温度25 ℃,转速125 r/min),培养40 h,每隔2 h取样.测其OD600监测生物锰氧化物的形成过程.取上层清液测铁锰离子质量浓度,考察BioMnOx形成过程对Fe(III)吸附的影响.
1.3 BioMnOx的制备
配置一批培养基,以相同的条件灭菌冷却,将母液以1%的接种量接入新配置的培养基中,将其置于恒温振荡器中培养(温度25 ℃,转速125 r/min)40 h左右生成黑色颗粒沉淀,即培养完全.将培养完全的培养基转入灭菌的离心管中,置于高速离心机中离心20 min(温度25 ℃,转速5 000 r/min),弃掉上清液用无菌水反复水洗3次,反复离心,得到BioMnOx.
1.4 BioMnOx的形成过程中Fe(III)吸附及APAP氧化相互影响
配置一定量的培养基,加入不同质量浓度的APAP溶液(10,100 mg/L)、Fe(III)以柠檬酸铁铵形式加入(1 g/L)以及1%的恶臭假单胞菌母液,同时设置3组平行样.将Pseudomonasputida接入的时间记为零时刻,每隔一定时间取样.通过HPLC和ICP-OES测定溶液中不同时刻APAP的剩余质量浓度和Fe(III)质量浓度,考察BioMnOx形成过程中Fe(III)吸附及APAP氧化相互影响.
另外,配置不同Mn2+质量浓度(8.13,16.25,24.38,32.50 mg/L)的培养基,接种1%的Pseudomonasputida母液,每组设置3个平行样.不同量的BioMnOx生成后离心收集,加入APAP溶液中,将BioMnOx接入的时间记为零时刻,每隔一定时间取样,检测剩余APAP质量浓度,以此探讨BioMnOx的量对APAP氧化的影响.
1.5 Pseudomonas putida对APAP的氧化活性
制备6组相同条件下生成的BioMnOx,振荡离心后弃掉上清液,将BioMnOx与10 mg/L APAP的无菌溶液混合,同时向其中3瓶加入50 mg/L的叠氮化钠,作为抑制组1,其余3瓶不加作为对照组1[9].将其置于恒温振荡器中振荡(温度 25 ℃;转速 125 r/min).将加入BioMnOx的时间记为零时刻,在振荡2 h及24 h时各取一次样,测定溶液中APAP质量浓度.通过两组APAP去除情况的比较,探讨Pseudomonasputida的氧化活性对Fe(III)吸附及APAP氧化的影响.
振荡24 h后,将两组溶液分别超声1 h,取其上清液检测溶液中铁锰质量浓度,探讨其在APAP降解过程中的作用.
1.6 降解产物分析
配置一定质量浓度的APAP溶液,加入生成的BioMnOx,将BioMnOx接入的时间记为零时刻,每隔一定时间取样,通过GC-MS系统分析降解产物.
1.7 分析方法
1.7.1 样品分析
实验中不同时刻所取的样品于20 ℃、4 000 r/min条件下离心20 min,上清液经0.22 μm滤膜过滤后于4 ℃下保存,用于后续实验分析.
APAP质量浓度通过HPLC-UV系统分析,配置反向C18高效液相色谱柱(150×4.6 mm,5 μm).柱温设为30 ℃,进样量20 μL.流动相,V(甲醇)∶V(超纯水)(含0.05%冰乙酸)=20∶80,流速0.5 mL/min,检测波长240 nm[9].
溶液中铁锰离子质量浓度通过电感耦合等离子体发射光谱仪(ICP-OES)测定.
1.7.2 动力学实验
对APAP的降解、Mn2+的氧化过程进行一级反应动力学拟合,反应速率方程如下
(1)
积分形式为
(2)
式中:t为时间,h;ρ为t时刻APAP或Mn2+的质量浓度,mg/L;ρ0为0时刻APAP或Mn2+的质量浓度,mg/L;K为反应速率常数.
1.7.3 降解产物分析
采用GC-MS分析主要降解产物,GC-MS条件[20]:进样口温度280 ℃,采用程序升温,初始60 ℃,保持3 min,然后以10 ℃/min升至280 ℃,保持3 min;He为载气,流量1 mL/min,分流20 mL/min,进样量1 μL;EI源为电离源,70 eV.样品前处理:取水样150 μL,加入气相色谱瓶中,以轻柔的氮气吹干,然后加入150 μL无水四氢呋喃和60 μL BSTFA,加盖密封后置于40 ℃烘箱中衍生30 min,用于GC-MS分析.
1.7.4 BioMnOx表征方法
分别收集培养24,44 h时的BioMnOx,将其置于冷冻干燥机中冷冻干燥,分别取少许置于电镜样品台上作喷金处理,然后采用扫描电镜(SEM,Quanta FEG 250,美国)、透射电镜(TEM,Talos F200 X,美国)对BioMnOx的微观结构进行观察.
2 结果与讨论
2.1 BioMnOx的形成过程
从P.putidaQSY-1接种至6 h,培养基保持澄清透明,颜色为橙黄色,从6 h起培养基溶液颜色加深,为红棕色,观测到10 h无明显沉淀,但溶液逐渐浑浊;从10 h起开始逐渐出现棕黑色锰沉淀物,到20 h之间培养基呈黑褐色胶体状溶液,静置不分层;20 h后大量絮状棕黑色沉淀逐渐积累,静置后固液边界清晰,上清液澄清透明;由图2可知,OD600也与观测现象吻合,在6~18 h呈线性递增,20 h后OD600几乎不再增加,但误差棒增大,是因为锰氧化物颗粒变化较大,培养基由乳浊液变化至可分离悬浊态,说明BioMnOx在形成过程中自身结构发生变化.
BioMnOx生成过程中的微观结构变化见图1.可以看出,在BioMnOx形成初期,BioMnOx表面较为平整,但内部嵌布颗粒,粒径较小,直径约为49.9 nm,可见此时生物氧化形成的锰氧化物结晶较弱[21],由TEM图可以看出,BioMnOx为无定型的纳米颗粒.在24~44 h,BioMnOx结构发生了变化,由平整演变为密集、突出的颗粒状,颗粒边界更为清晰,粒径约70 nm.Tebo等研究发现生物锰氧化物初级产物的晶体结构会随时间发生变化,在初级产物中共存着正六边形和假直角型两种晶体结构.12 h时正六边形结构占主导,之后假直角结构逐渐变成主导[22].
由图2可知,溶解性锰离子在0~10 h质量浓度变化较小,10~20 h迅速降低,从44.44 mg/L降至0.86 mg/L,尤其是在14~16 h,2 h内减少了40.8 mg/L.Mn离子的急剧减少并非是Mn离子的完全氧化,因为OD600在2 h内没有急剧增加,仍处于线性递增,Mn离子的急剧减少应源于BioMnOx对Mn离子的吸附.Forrez等[23]观察到锰氧化物形成过程中表面可吸附大量的Mn离子,培养20 h时Mn2+/Mn4+的比率为0.57 mg/mg.20 h后,Mn离子质量浓度再次处于缓慢降低的过程,最终Mn离子质量浓度稳定在0.15 mg/L左右.
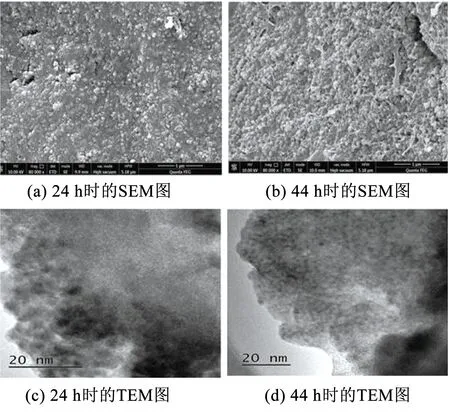
图1 不同时刻BioMnOx的TEM结构

图2 锰氧化物的形成
2.2 BioMnOx的形成过程对Fe(III)的吸附及对APAP的氧化
由图3可知,BioMnOx对Fe(III)的吸附发生在对APAP的氧化之前.在APAP的共存体系中,无论APAP是低或高质量浓度(10,100 mg/L),Fe(III)的质量浓度变化趋势一致,即0~12 h质量浓度变化不大,保持在123 mg/L左右,而12~16 h急剧下降至1.3 mg/L左右,在20 h后少量回增又下降至稳定,保持在1.2 mg/L左右.12~16 h是锰氧化物初级产物形成期,其结构正逐渐由正六边形演化至假直角型[22],Tebo等[22]研究发现后者中的Mn(Ⅳ)缺位较少,而Mn(Ⅳ)空缺位点的数量直接影响到吸附能力.因此,在12~16 h BioMnOx对Fe(III)显示出较强的吸附性能,16~20 h则保持稳定.20 h以后APAP开始被BioMnOx氧化,部分Mn(IV)氧化物作为电子受体转化为溶解性的Mn(II),结构改变致使吸附位点减少,已吸附的Fe(III)被解吸释放.通过比较10及100 mg/L的APAP体系可知,在100 mg/L体系中Fe(III)的释放量(22.4 mg/L)几乎为10 mg/L体系中(2.8 mg/L)的10倍.吸附后期随着P.putida的氧化作用,持续地将Mn2+转化为BioMnOx,继而对释放的Fe(III)重新吸附至接近1.2 mg/L.因此,APAP与Fe(III)共存时,BioMnOx对APAP的氧化影响了对Fe(III)的吸附,致使其发生解吸,但解吸可通过持续的BioMnOx生成而消除.

图3 不同初始质量浓度APAP与Fe3+、Mn2+质量浓度随时间的变化

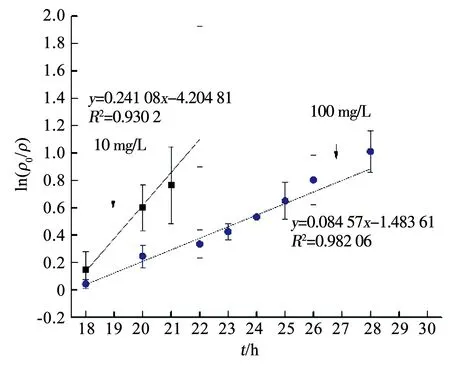
图4 降解速率曲线

表1 APAP降解及锰氧化过程一级动力学方程参数
APAP的存在对Mn2+的变化规律亦产生了影响.由图5可知,在只有Fe3+存在而无APAP时,Mn2+在14~16 h由38.2 mg/L急剧减少至3.62 mg/L,然而在APAP存在时,Mn2+减少速率放缓(图3).为分析APAP质量浓度对Mn2+氧化速率的影响,取图3(a)中18~23 h、图3(b)中18~30 h Mn2+的氧化速率进行一级反应动力学拟合,反应速率方程拟合曲线如图6所示,可以看出,Mn2+的氧化速率符合一级动力学方程,且速率常数与相对应质量浓度的APAP氧化速率常数接近,即10,100 mg/L APAP质量浓度下的Mn2+氧化速率常数KMn分别为0.760 55和0.134 36 h-1,K(Mn,10)远大于K(Mn,100),同时K(APAP,10)也远大于K(APAP,100)(表1),表明100 mg/L质量浓度下的Mn2+氧化速率低,导致该质量浓度下APAP的降解速率较慢.锰氧化速率的降低应该是由APAP对Pseudomonasputida生长抑制作用导致,本课题组前期实验表明,高质量浓度下(100 mg/L)的APAP对Pseudomonasputida生长有抑制作用,100 mg/L的APAP培养基中细菌数量较10 mg/L减少了约75%,因此,对Mn2+的氧化速率产生影响,进而导致高质量浓度APAP降解速率降低.

图5 无APAP存在时铁锰离子质量浓度变化

图6 Mn2+氧化速率曲线
在Pseudomonasputida氧化Mn2+生成BioMnOx过程中,虽然对Fe(III)的吸附先于对APAP的氧化,但对Fe(III)的吸附并未对APAP的氧化速率产生影响;APAP的氧化过程中则会导致Fe(III)解吸的发生,但新增的BioMnOx可对Fe(III)再吸附;APAP质量浓度增加可抑制Mn2+的氧化速率来减缓APAP的降解速率.因此,Pseudomonasputida对Mn2+的氧化活性及BioMnOx的量将会对APAP降解造成影响.
2.3 Pseudomonas putida的氧化活性对Fe(III)吸附及APAP氧化的影响
本实验利用叠氮化钠抑制Pseudomonasputida的活性[9].由图7可知,Pseudomonasputida的活性被抑制后APAP的降解效果变差,反应2 h时,对照组1中APAP的去除率为72%,而抑制组1中其去除率只有33%,反应24 h时,对照组1中APAP的去除率达100%,实现完全降解,而抑制组1中APAP的去除率与反应2 h时相比几乎没有变化,说明PseudomonasputidaMn2+氧化活性的抑制严重影响了APAP的降解.Sabirova等[24]也发现了叠氮化钠可以抑制Pseudomonasputida的活性使其降解17α-炔雌醇的能力变差,这是因为由于叠氮化钠的抑制作用,Pseudomonasputida活性受到抑制后就无法继续氧化培养基中的Mn2+,使得培养基丧失持续产生BioMnOx的能力,进而导致有机污染物不能被完全降解.所以,抑制组1中APAP的去除率没有随反应时间的延长而有所变化.抑制组1中反应2 h时达到的33%的去除率可能是加入叠氮化钠前培养基中已经生成的BioMnOx作用的结果.以上证明APAP质量浓度的增加可以抑制培养基中Mn2+的氧化速率进而减缓APAP的去除速率.

图7 APAP降解效率
对照组1和抑制组1超声后检测溶液中溶解性铁锰离子质量浓度,结果显示两组中铁离子质量浓度均非常低,约为零,这表明Pseudomonasputida的氧化活性抑制对BioMnOx吸附没有产生影响.抑制组1中Mn2+质量浓度平均为9.3 mg/L,对照组1中Mn2+质量浓度仅为0.5 mg/L.这也表明BioMnOx对Fe(III)的吸附并非为与Mn2+的离子交换机制.结合上文实验结果,对照组1中APAP的去除率达100%,而抑制组1中APAP的去除率仅达33%,这说明培养基中Mn2+的持续氧化是实现APAP能够达到完全降解的重要因素.对照组中由于BioMnOx表面负载少量Mn2+,在与APAP溶液混合后,表面的Mn2+不断被释放出来,Pseudomonasputida持续氧化Mn2+,继而生成新的BioMnOx,不断生成的BioMnOx能够促使溶液中的APAP持续降解,所以,对照组1中的APAP去除效果较抑制组1更为显著,同时,由于培养基中Mn2+被不断消耗生成新的BioMnOx,其超声后的质量浓度较低.而抑制组1中,由于Pseudomonasputida的活性被抑制,进而Mn2+氧化作用被抑制,不能生成新的BioMnOx,APAP的降解只能由培养基中原有的BioMnOx来实现,由于原有的BioMnOx量很少,抑制组1中的APAP不能实现完全降解,超声后的Mn2+质量浓度也比对照组1中的高.因此,BioMnOx量对APAP的氧化产生一定影响.
2.4 BioMnOx量对APAP氧化的影响
BioMnOx量对APAP氧化速率常数的线性关系采用一级反应动力学进行拟合(见图8和表2),其相关系数均较好(R2=0.92~0.99),符合一级反应动力学模型.Mn2+质量浓度为24.38,32.50 mg/L时所生成的BioMnOx能够在14 h内实现APAP降解完全,而Mn2+质量浓度为8.13,16.25 mg/L时所生成的BioMnOx分别在24,18 h时才能实现APAP降解完全,说明APAP所需完全降解的时间与生成的BioMnOx的量成正相关.

图8 速率常数曲线

表2 速率常数曲线参数
2.5 APAP的氧化降解途径推断
采用GC-MS对APAP氧化降解的中间产物进行分析[12],结果列于表3,根据表3推测APAP氧化降解可能的降解途径,结果见图9.
首先APAP苯环上的乙酰基被取代生成乙酰胺(1)、对氨基苯酚和对苯二酚(6),(1)进一步被氧化为乙醇酸(2),继而生成乙二酸(3),最终氧化为二氧化碳和水;同时对氨基苯酚的-NH2被取代生成(6),之后(6)脱下羟基生成苯醌(7),(7)进一步开环裂解、双键打开生成顺丁烯二酸,顺丁烯二酸继续被氧化生成(3)[25],最终被矿化为二氧化碳和水,或者对氨基苯酚被氧化生成苯醌亚胺(4),进一步再生成(7),然后重复上述降解途径;(6)同时被氧化生成(7)和丁二酸(5),(5)继续氧化生成(3),而(7)重复上述降解途径,最终生成二氧化碳和水.

表3 APAP降解的中间产物结构推测

图9 APAP氧化降解可能的降解途径
3 结 论
1)BioMnOx为无定型的纳米颗粒,在形成过程中自身结构发生变化,表面由平整变为密集、突出的颗粒状,颗粒边界更为清晰,颗粒粒径从约49.9 nm长至约70 nm.BioMnOx在形成过程中Mn离子的急剧减少主要是由于BioMnOx对Mn离子的吸附.
2)BioMnOx形成过程中对Fe(III)的吸附发生在对APAP的氧化之前,但对Fe(III)的吸附并未对APAP的氧化速率产生显著影响.APAP氧化降解过程以及不同APAP质量浓度对Mn2+氧化速率影响过程均符合一级动力学方程.
3)APAP质量浓度增加可抑制Mn2+的氧化速率来减缓APAP的降解速率.Pseudomonasputida对Mn2+的氧化活性及BioMnOx的量对APAP降解有一定的影响.
4)在APAP氧化降解过程中,生成了对苯二酚、乙酰胺、对氨基苯酚等一系列产物,随后继续被氧化为结构更简单的有机物,最后生成CO2和H2O,实现APAP的降解.
猜你喜欢
陶瓷学报(2021年4期)2021-10-14
陶瓷学报(2021年1期)2021-04-13
食品安全导刊(2020年21期)2020-12-03
食品安全导刊(2020年18期)2020-12-03
湖南农业科学(2020年1期)2020-04-18
中学生数理化·高一版(2016年7期)2016-12-07
山东工业技术(2016年15期)2016-12-01
试题与研究·中考化学(2016年1期)2016-09-30
中学化学(2016年4期)2016-05-30
中学化学(2015年9期)2016-04-14
