
1例软骨-毛发发育不全的临床及遗传学特征分析
2021-03-19 08:24姜文君张惠文
上海交通大学学报(医学版) 2021年2期
杨 奕,姜文君,张惠文
上海交通大学医学院附属新华医院上海市儿科医学研究所小儿内分泌与遗传代谢病研究室,上海200092
软骨- 毛发发育不全(cartilage-hair hypoplasia,CHH,OMIM #250250) 由McKusick 于1965 年 首 次 报道[1],也被称为McKusick 型干骺端软骨发育不全或短肢侏儒免疫缺陷症,为一种罕见的常染色体隐性遗传性疾病。CHH 已经在多个国家陆续被报道,目前在阿米什人及芬兰人中发病率最高,分别为1/1 000~2/1 000[2]和1/23 000[3]。
该病是由于线粒体处理RNA的内切酶复合物RNA组分(RNA component of mitochondrial RNA processing endoribonuclease,RMRP)基因突变导致。RMRP 基因编码MRP 核糖核酸酶的RNA 组分,为一非编码RNA[4]。目前100余种RMRP 基因致病突变位点被相继报道,最常见的致病突变是g.70A→G,在阿米什人中发生率为100%,芬兰人中为92%[5-6]。临床主要表现为身材矮小、四肢短小且与躯干不成比例、毛发细软稀疏、骨骺端软骨发育不良等;此外,多伴有多系统异常,如不同程度的免疫缺陷、再生不良性贫血、恶性肿瘤、先天性巨结肠、精子形成异常、皮肤及内脏肉芽肿等其他临床表现也被相继报道[2]。该病临床表现差异显著,且缺乏特异性,早期诊断困难。
CHH 在我国的发病率尚不清楚。1995 年国内报道2例,为家系同胞兄弟,2 人均以短肢性侏儒、双下肢畸形、毛发色淡为主要临床表现,且伴有鱼鳞状皮损和智力低下,未进行基因检测[7]。目前,国内鲜有该病的报道。不排除该病临床表现多样、临床医师认识不足的可能。目前,已知基因的全外显子测序是诊断疑难杂症、罕见病的常规方法;但是RMRP 基因为非编码RNA,没有外显子,不在探针列表。因此,该病若选择使用全外显子测序检测,找不到突变位点。目前进行CHH 诊断,必须在临床疑似基础上,选择第一代测序技术,即进行RMRP 基因Sanger 测序,方可检出致病变异。本文对1 个CHH家系进行临床表型及遗传学特点分析。
1 临床资料
1.1 患儿基本情况
先证者,女,4 岁4 个月,因“自幼身高增长缓慢及双下肢较短”就诊。患儿系G3P1,足月剖宫产(孕母妊娠期高血压),出生体质量3.1 kg,出生身长具体不详,否认出生窒息及抢救史,父母非近亲结婚。母孕期多次超声检查提示四肢偏短,具体不详。生后喂养可,按时添加辅食,自幼身高增长缓慢,毛发稀少且生长缓慢。3月龄体检示身高落后,13.3月龄身高为65.5 cm(<-3SD),体质量为7.5 kg(<-2SD),明显落后于同龄儿。体格生长记录见图1。当时于外院行数字化X 线片检查提示尺骨远端杯口样改变,胸椎、腰椎生理曲度存在,形态可,未见明显异常改变。头颅磁共振检查未见明显异常。运动、语言及智力发育正常。父亲身高178 cm,母亲身高163 cm,父亲幼时有“O”形腿表现,无家族遗传性疾病。
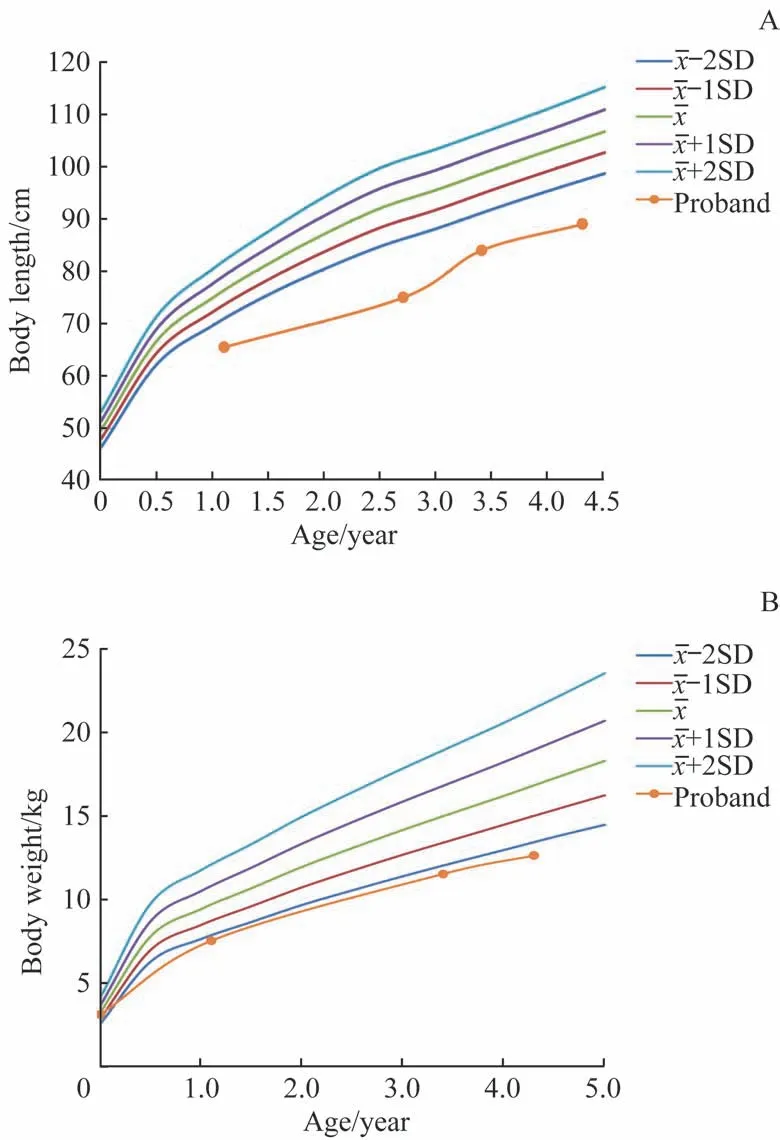
图1 先证者身高(A)及体质量(B)记录Fig 1 Body length(A) and body weight(B) records of the proband
体格检查:身高89 cm (< -3SD),坐高52 cm(< -2SD),指间距77.5 cm,体质量12.6 kg(< -2SD)。神志清楚,精神反应可。面容无特殊,毛发稀疏、色浅,步态正常,四肢偏短,胸廓无突出,肋缘无外翻,膝关节无外翻,脊柱畸形(-),臀部后翘。背部少许蒙古斑。心、肺、腹未见明显异常,神经系统查体阴性。辅助检查:血甲状腺激素、甲状旁腺激素、血钙、血镁、总25-羟维生素D 及碱性磷酸酶水平未见明显异常,血磷1.34 mmol/L,胰岛素样生长因子-1(insulin-like growth factor-1,IGF-1)103 ng/mL,胰岛素样生长因子结合蛋白3(insulin-like growth factor binding protein-3, IGFBP-3)4.87 μg/mL。左手X 线片提示骨龄5 岁(图2A)。下肢正侧位数字化X 线片提示右股骨下端及右胫骨两侧干骺端密度偏高,周围软组织未见明显异常(图2B~D)。胸腰椎正侧位X 线片提示胸腰椎生理曲度存在,未见侧弯及滑脱,诸椎体及附件形态尚可,骨质未见明显异常,诸椎间隙未见明显狭窄(图2E、F)。
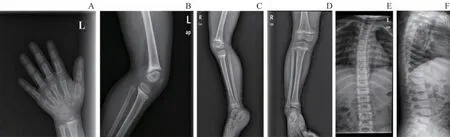
图2 先证者影像学检查Fig 2 Radiographs of the proband
1.2 基因突变检测方法
1.2.1 样本DNA 的制备 在获得知情同意后抽取患儿及父母静脉血2 mL,用乙二胺四乙酸二钠(EDTA-Na2)抗凝。用血液基因组DNA 提取试剂盒(Omega,美国)提取全基因组DNA。
1.2.2 全外显子基因测序分析 首先使用HiSeq 2500 高通量模式测序,按HiSeq 2500 标准流程进行。先将上述步骤中提取的DNA 片段化。制备文库。目标序列测序覆盖度不低于99%。用探针捕获遗传性疾病panel 的外显子及邻近区域,最后送高通量测序平台HiSeq 2500(Illumina,美国)进行测序。对原始数据进行过滤,得到高质量的测序结果并进行分析。应用MaxEntScan 软件对剪切位点变异进行危害性预测。变异位点经单核苷酸多 态 性 数 据 库 (database of single nucleotide polymorphisms,dbSNP)、千人基因组数据库、外显子组整合数据库(Exome Aggregation Consortium,ExAC)、外显子组测序项目(Exome Sequencing Project,ESP)等频率数据库及人类基因变异数据库(human gene mutation database,HGMD)、NCBI 临 床 突 变 数 据 库(ClinVar)等疾病数据库进行关联注释及疾病风险评估。
1.2.3 一代测序(Sanger 法)检测 采用直接扩增先证者RMRP 基因及父母验证的方法,并将扩增结果进行测序。分析测序结果并与标准序列(序列号:NG_017041.1)比对,寻找突变位点。
1.3 基因测序结果
该家系进行全外显子基因测序分析后,结果提示COL10A1,c.2T→C 基因杂合变异,该位点变异来源于父亲。该基因变异可引起脊柱骨骺发育不良,主要累及脊柱骨骺和长骨末端骨骺致进行性骨软骨发育不良。该病遗传模式为常染色体显性遗传。若该位点变异有致病性,先证者父亲应有骨病表现。但是父亲身高正常,因此推断该位点为罕见良性变异。同时检测出以下基因变异:HERC2(c. 9939C→T, c. 7231_7245delTGCCAGTGCTTGGCT,常染色体隐性遗传);FAM111A(c.1628C→A,常染色体显性遗传);LRP5(c.1395A→C,常染色体显性遗传);CPS1(c.213C→T,c.195C→T,常染色体隐性遗传);KLHL24(c.1702G→T,常染色体显性遗传);但因基因型与临床表型不相符或致病性弱,予排除。
进一步分析临床特征,患儿头发稀疏,身材显著矮小且不对称,考虑CHH,因此进行RMRP 基因一代测序(Sanger 法)检测。检测发现,先证者RMRP 基因存在复合杂合变异,基因型为g.181G→A/g.255C→T。前者来源于父亲,已有相关文献[6]报道;后者来源于母亲,根据美国医学遗传学与基因组学学会(American College of Medical Genetics,ACMG)指南[8],从突变频率、遗传方式、家系分离及表型高度相符等方面进行评级,结果为可能致病(likely pathogenic,PM2+PM3+PP1+PP4-M),未在HGMD报道过,为新发现的突变(图3)。
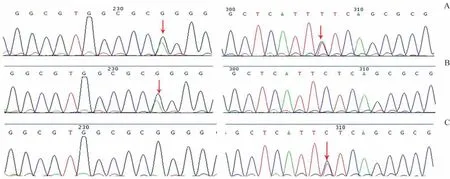
图3 家系RMRP基因Sanger测序峰图Fig 3 RMRP gene Sanger sequencing of the family
2 讨论
本研究分析了1例CHH 患儿的临床表现。患儿运动、语言及智力发育正常,以生长落后、非匀称性矮小、四肢偏短、毛发稀疏为主要症状,这与CHH 最常累及肌肉骨骼系统有关。典型的CHH 患者出生身长常小于平均值,在生后2年内即出现身高增加缓慢,青春期生长加速可能很小甚至无[9]。既往相关文献[10]报道,CHH 男性患者成年中位数身高为131.1 cm(110.7~149.0 cm),女性患者成年中位数身高为122.5 cm(103.7~137.4 cm)。
患儿有毛发生长异常及形态学改变,表现为纤细、稀疏、色浅,“丝质”发,容易折断。van der Burgt 等[11]学者研究发现CHH 患儿毛发微观结构发生改变,毛干直径变小,为正常毛发直径的50%~60%,成分改变,拉伸试验提示比正常毛发脆弱、易断。
本病例影像学检查均提示骨发育异常。以累及干骺端及骨骺为特点,最常累及双下肢,表现为干骺端出现齿状边缘,呈喇叭口样、硬化[12],且干骺端异常的程度与生长速度及最终身高相对应。临床病例研究[13]表明,尺骨和桡骨长度短于肱骨,股骨长度短于胫骨,胫骨比腓骨受累更严重。CHH 患者中轴骨大小也受累,提示该综合征中存在全身骨骼生长功能障碍。该病还可有腕关节或踝关节的关节松弛、肘部伸展受限、腰椎前凸、踝关节内翻、膝关节内翻、脊柱侧凸等改变;其中腰椎前凸加重、韧带松弛和脊柱侧凸常见,并且与年龄有关[13]。本研究中CHH 患儿仅有上述部分特征性相关改变,考虑可能与发病年龄及病程有关。
CHH患者临床表现多样化,可累及多器官、多系统,骨骼影像学改变不典型,实验室检测无特异性指标,诊断较困难,目前尚无统一标准。现临床诊断主要依据四肢非均匀性矮小、毛发细软稀疏等典型的临床表现以及骨骺软骨发育异常的影像学特征,伴有免疫缺陷、大红细胞性贫血以及消化系统异常等其他症状可提示诊断。对于临床特征不明显的患者,需与临床表现相似的疾病进行鉴别诊断,并通过RMRP 基因突变分析明确诊断并指导产前诊断。
RMRP 基因位于染色体9p13,无内含子及外显子,转录本包括3 个启动子——邻近序列元件(proximal sequence element,PSE)、TATA盒、转录因子结合位点上游的转录起始位点[4,14]。该基因产生长度为268 bp 的非编码RNA,无外显子,不编码蛋白质,不在常见全外显子检测试剂盒的探针列表。本病若使用全外显子测序检测,找不到致病性基因突变位点。对于疑似CHH 的临床病例,在行基因检查时需针对性进行RMRP 一代测序(Sanger 法)检测,且测序范围应包含转录区和启动子区等整条基因。
RMRP基因致病突变多数发生在编码区,如本例患儿RMRP基因复合杂合变异,基因型为g.181G→A/g.255C→T,前者来源于父亲,后者来源于母亲。突变g.255C→T为新发现,丰富了该疾病基因突变谱。RMRP基因在启动子、转录区及终止密码子后的突变也有相关报道[15]。RMRP 致病突变可导致线粒体RNA 内切酶活性降低,引起rRNA与mRNA裂解减少,核糖体组装及细胞周期调控发生障碍[16-17]。rRNA 裂解受损与骨发育不良的严重程度密切相关,mRNA 裂解减少与毛发发育不全、免疫缺陷、血液学异常改变及罹患癌症的风险增加有关[15,18],因此临床上可呈现多系统异常表现。本研究中的患儿尚未出现免疫系统、血液系统及肿瘤病变,需要进一步的专科随访。
有些疾病的临床表现与CHH 相似,鉴别诊断包括其他常染色体隐性遗传性骨骼发育不良以及矮小症(可能伴有免疫功能缺陷),如不伴稀毛症的干骺端发育不良(OMIM #250460)、严重骨骼发育不良改变(OMIM#607095) 及Shwachman-Diamond 综 合 征(OMIM #260400)[15]。
目前,尚未发现针对CHH 疾病的特效疗法。一般采用对症治疗手段。针对矮小症患儿,重组人生长激素的治疗目前尚存在争议[19-20],因其可能增加罹患肿瘤的风险。针对反复严重感染或合并严重联合免疫缺陷的患儿,可行骨髓移植或造血干细胞移植治疗[21]。本研究患儿尚未出现贫血、严重骨骼畸形、内分泌异常及肿瘤改变。为更科学诊治CHH 患儿,应建立专科随访制度。不伴有多系统异常的患儿,每1~2 年至少随访1 次,随访内容应根据患儿年龄及病情严重程度制定。专科随访时进行体格检查、营养状况评估,行骨龄及肢关节X 线检查评估骨骼发育情况。若合并内分泌系统异常,建议每3个月门诊随访,评估身高线性生长情况、身材比例及青春期发育情况;对于使用重组人生长激素治疗的患儿,同时应完善肝功能、血糖、血尿常规、甲状腺功能、IGF-1 等检验[20]。长期随访有助于及时发现患儿的临床并发症,并进行早期干预,改善疾病预后。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国生殖健康(2020年4期)2021-01-18
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
肿瘤预防与治疗(2019年6期)2019-07-30
中国生殖健康(2019年11期)2019-01-07
青少年科技博览(中学版)(2017年5期)2018-02-28
百科知识(2015年18期)2015-09-10
小火炬·阅读作文(2014年5期)2015-04-07
湖北农业科学(2014年11期)2014-09-10
