
Mn在γ′-Ni3Al中的择优占位
2021-03-13 13:01黄仁忠刘丽丽魏媛媛刘雅娟
沈阳师范大学学报(自然科学版) 2021年6期
黄仁忠, 刘丽丽, 魏媛媛, 刘雅娟
(沈阳师范大学 物理科学与技术学院, 沈阳 110034)
0 引 言
镍基高温合金作为一种具有优异力学性能的高温结构材料,在高温领域得到了广泛的应用。这种高温合金的关键组织是合金Ni3Al(γ′相)。γ′-Ni3Al具有异常的屈服应力行为和优良的高温力学强度。其在镍基合金中体积分数高达近70%[1],是用于高温应用的商用镍基高温合金的关键强化成分[2-3]。研究表明,添加第三元取代元素可以提高γ′-Ni3Al的力学性能和高温抗氧化性[4]。在Ni3Al中,第三元取代元素可以独占Al亚晶格、Ni亚晶格,或同时占据Ni,Al 2种晶格。因此,想要研究替代元素对Ni3Al力学性能的作用,首先要清楚替代元素在Ni3Al中的占位行为。
探索替代元素在Ni3Al中的占位行为对理解和研究其对Ni3Al力学性能的强化作用十分重要。实验上,已有多种研究手段探索合金化元素在Ni3Al中的占位问题,如原子探针[5]、透射电子显微镜[6]、X射线衍射等[7]。但是,由于实验研究成本较高,只有少数的金属元素能够被重点研究。综合来看,理论研究方法可以很好地满足元素占位研究的要求。早期Freeman研究组[8]就利用第一性原理的方法研究了合金化元素在Ni3Al中的占位问题。2004年,Wang等[9]也利用第一性原理方法探究了铂族元素在Ni3Al中的占位。Ruban等[10-11]介绍了利用转移能来预测合金化元素在0 K时在Ni3Al中的占据位置优先性的方法。在诸多第三元取代元素中,发现关于Mn在Ni3Al中的占位具有较大争议。Sluiter等[12]利用基于局域密度近似的电子总能计算,得出了Ni3Al中Mn占据弱Al亚晶格的结论。Gleeson[13]将第一性原理与Wagner-Schottky模型结合得出Mn在Ni3Al中占位情况随温度及成分变化的结论。Ochial等[14]通过溶解度实验研究认为Mn在Ni3Al中没有明显的占据倾向。
本文将通过3种方法来研究Mn在Ni3Al中的占位情况。第一种为通过掺杂原子的缺陷形成焓来判断其占位情况。第二种通过掺杂原子占据主相中某一类亚晶格的位置,以及被替代原子占据另外一类亚晶格位置的转移能来判断其占据倾向。第三种则是利用Wagner-Schottky模型来探究Mn在Ni3Al中的占据倾向并探究其与合金成分及温度的关系。
1 计算设置及模型
研究使用Castep[15]软件中基于密度泛函理论(DFT)[16]的第一性原理平面波赝势方法。相互关联能的处理全部采用广义梯度近似(GGA)中的PBE形式[17](势函数采用倒空间表述的超软赝势)。在进行结构计算时,布里渊区的k点选用Monkhorst-Pack的取样方式[18],平面波截断能均设置为300 eV,k点网格选为3×3×3。所有结构自洽收敛条件均设为:体系相邻两步弛豫的能量差小于1.0×10-6eV,每个原子上的最大作用力小于0.03 eV/Å,应力偏差小于0.05 GPa,原子位移小于1.0×10-3Å。
首先建立2个由32个原子组成的无缺陷2×2×2的Ni3Al超胞结构,结构中心分别是Ni和Al原子,如图1所示。在Ni3Al超晶胞基础上进行Mn原子置换,替代原子以及空位可占据结构中心的Ni原子或Al原子,形成缺陷结构。考虑到周围局部原子弛豫对晶格常数的影响,对所有构型进行几何优化,以使结构中所有原子及晶格常数完全弛豫。计算中考虑磁性的影响。
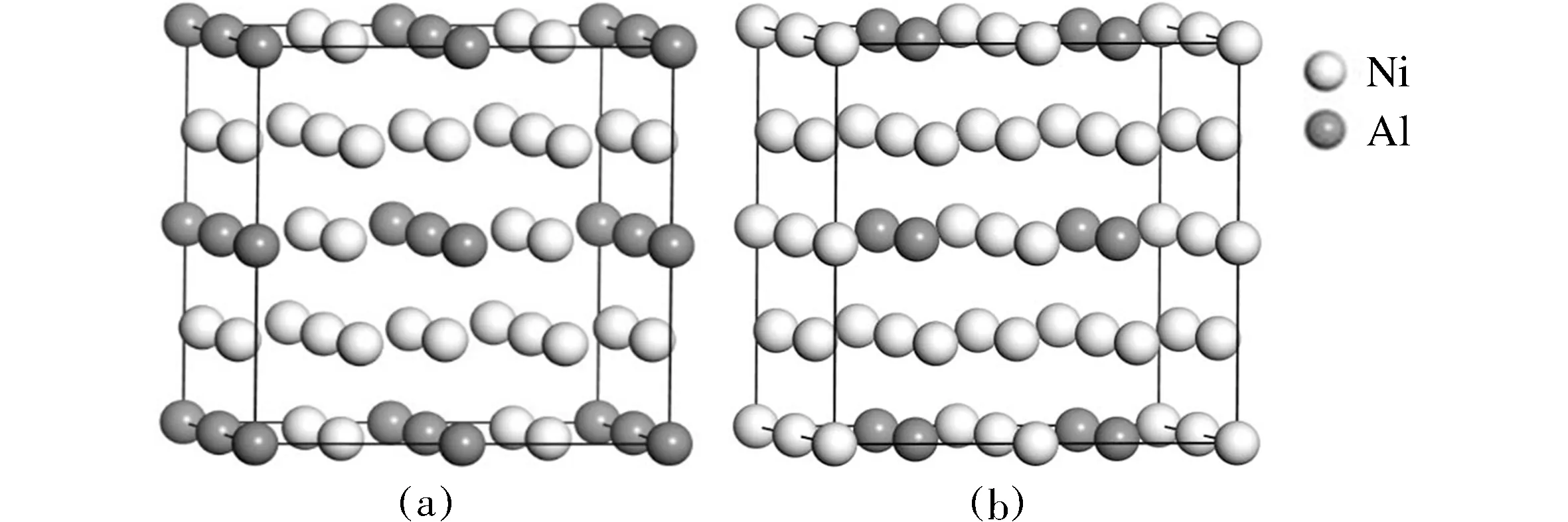
图1 (a) 以Al为中心的Ni3Al超胞; (b) 以Ni为中心的Ni3Al超胞Fig.1 (a) The Ni3Al supercell centered on Al; (b)The Ni3Al supercell centered on Ni
2 结果与讨论
对各种超晶格结构进行弛豫,以优化纯Ni3Al超晶胞和含缺陷的超晶胞的晶格参数,见表1。计算得到Ni3Al晶格常数为3.588 Å,与实验值[19]及其他计算结果[20]非常接近。因此,在这种精度下,可以进行Mn原子在Ni3Al中占据倾向的研究。另外,从表1中可以清楚地看出,与纯超胞相比,Mn原子加入会使晶胞变大,且对于同浓度的Mn原子的占据,Ni23Al8Mn超胞的晶格参数大于对应的Ni24Al7Mn超胞的晶格参数。Mn原子替代Ni原子结构的体积变化要比Mn原子替代Al原子的更为明显。这是由于Mn原子半径与Al原子半径相差较小,与Ni原子半径相差较大的缘故。
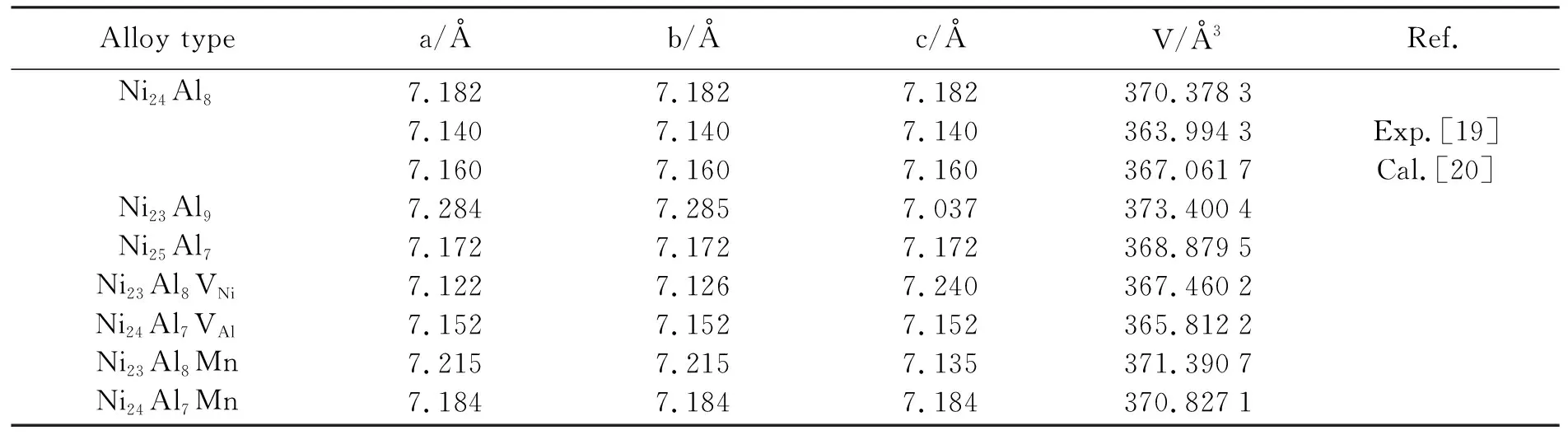
表1 Ni3Al超晶胞以及各缺陷结构平衡晶格常数及体积Table 1 The relaxed lattice constants and volumes of Ni3Al supercell and its defect structures
对于纯Ni3Al,计算得到的每单位Ni3Al晶胞的磁矩为0.82μB(表2),与文献计算结果基本一致[21-22]。

表2 Ni3Al磁矩(μB/unit)计算结果Table 2 Calculated magnetic moment of Ni3Al (μB/unit)
2.1 利用位置优先能判断Mn在Ni3Al中的占位
Mn在Ni3Al中的取代能可通过位置优先能进行研究,位置优先能定义为2个形成能之间的能量差:[9]
Esite=ΔE1-ΔE2
(1)
其中ΔE1,ΔE2分别为Mn占据Al亚晶格和Mn占据Ni亚晶格的形成能[23]。形成能通常被定义为化合物和组成元素之间的能量差:
其中ENi,EAl和EMn分别表示纯体Ni, Al和Mn的单原子总能量。因此,可以从上述2种形成能推导出Esite的形式:
当Esite<0时,Mn倾向于占据Al亚晶格;当Esite>0时,Mn倾向于占据Ni亚晶格。
计算得到的Esite为1.449,大于0,所以在利用位置优先能判断Mn在Ni3Al中的占位情况时,可以明显看出Mn更倾向于占据Ni亚晶格,且较为强烈。
2.2 运用转移能判断Mn在Ni3Al中的占位
在Ni3Al中,假定Mn原子占据Ni亚晶格位置,而被替代的Ni原子则转移至Al亚晶格位置,相应的转移能定义为[10-11]
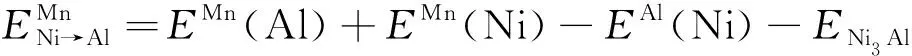
(5)
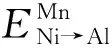
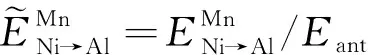
(6)
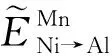
2.3 第一性原理结合Wagner-Schottky模型判断Mn在Ni3Al中的占据
2.3.1 0 K下Mn的占据倾向
在T=0 K时,Ni3Al中Mn的位置占据倾向仅由形成焓决定。缺陷形成焓的计算公式如下:
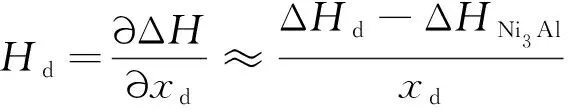
(7)
其中:ΔHNi3Al是无缺陷的32个原子Ni3Al超胞结构的形成焓;ΔHd为单个点缺陷在32个原子Ni3Al合金中的形成焓;xd为点缺陷的原子浓度; 空位缺陷浓度为1/31; 其他缺陷浓度为1/32。计算的各点缺陷形成焓见表3。

表3 Ni3Al合金中各点缺陷的形成焓Table 3 Defect formation enthalpies at various points in Ni3Al intermetallic compounds
Ni3Al中三元取代元素的占位一般可分为以下3种类型[24]:
1)HMnAl-HMnNi+HAlNi>HNiAl+HAlNi,在富Al,富Ni和标准化学计量比的Ni3Al中,尽管有Ni反位缺陷形成,X总是优先选择Ni亚晶格,即X表现出强烈的Ni位占据优先性。
2)HMnAl-HMnNi+HAlNi<0,在富Al,富Ni和标准化学计量比的Ni3Al中,即使已形成了Al反位,X总是优先选择Al亚晶格,即X表现出强烈的Al位占据优先性。
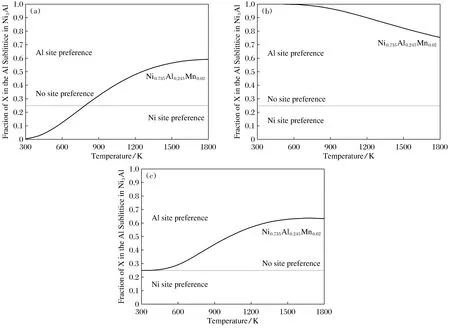
3) 0 由此可见,参数HMnAl-HMnNi+HAlNi已足以完全描述Ni3Al中任何三元元素在T=0 K的位点占据行为。当所有结构不考虑磁性,计算得到的HMnAl-HMnNi+HAlNi值为-0.066,小于0,Mn更倾向于占据Al亚晶格。考虑磁性影响后,HMnAl-HMnNi+HAlNi的值为0.343,而HNiAl+HAlNi=1.139,满足条件(3),所以在0 K温度下,在Ni3Al合金中Mn无明显占据倾向。与不加磁性的Ni3Al相比,Mn的占据有所改变,可见在探究Mn在Ni3Al中的占据行为时需考虑磁性的影响。 2.3.2 有限温度内Mn的占据倾向 上一部分中提出的分类仅在T=0 K时有效。在有限温度下,熵在决定Ni3Al中三元合金元素的位置占据倾向方面也起着重要作用。随着温度的升高,位置偏好可能发生变化。 图2为温度为1 273 K时,含1%Mn的Ni3Al中Mn占据Al亚晶格的摩尔分数随Ni摩尔浓度的变化关系。图2中水平虚线表示Al亚晶格和Ni亚晶格被随机占据,即没有占据倾向,此时在Ni3Al中Mn原子占据Ni亚晶格与占据Al亚晶格的比为3∶1。虚线以上部分代表替代原子倾向于占据Al亚晶格,虚线以下部分代表替代原子倾向于占据Ni亚晶格。从图2中可以明显看出, Mn一直都倾向于占据Al亚晶格,与合金成分无关。但相比于富Al区,在富Ni区Mn对Al亚晶格的占据倾向更为强烈。 图2 T=1 273 K时,Mn在NixAl0.99-xMn0.01中占据Al亚晶格摩尔分数与Ni浓度的关系Fig.2 The relation between the Molar fraction of Mn occupying the Al sublattice in NixAl0.99-xMn0.01and the concentration of Ni at T=1 273 K 接下来以富Al(Ni0.73Al0.25Mn0.02),富Ni(Ni0.75Al0.23Mn0.02)及标准化学计量比(Ni0.735Al0.245Mn0.02)的3种Ni3Al合金为例,探究温度对合金中Mn占据行为的影响。Mn占据Al亚晶格的分数随温度的变化如图3所示。从图3中可看出,在富Al的合金中,随着温度的升高,Mn的占据倾向由占据Ni亚晶格转变为占据Al亚晶格。在富Ni的合金中,整个温度范围内,Mn表现出强烈的占据Al亚晶格的倾向。在标准化学计量比的合金中,Mn的占据行为由无明显占据倾向转变为占据Al亚晶格。可见,无论是在富Al,富Ni,还是标准化学计量比Ni3Al中,Mn的占据倾向都受温度影响,尤其是富Al及标准化学计量比Ni3Al,Mn的占据晶格结果受到较大影响。 图3 富Al(a),富Ni(b)及标准化学计量比(c)的Ni3Al合金中Mn占据Al亚晶格的分数与温度的关系 本文使用3种方法探究了Mn在纯Ni3Al合金中的占据行为。得到的结果如下: 1) 利用位置优先能判断Mn的占据倾向。结果显示,Mn占据Ni亚晶格; 2) 利用转移能判断Mn的占据倾向。结果显示,Mn占据Ni亚晶格,但相对于位置优先能,占据倾向更弱; 3) 结合第一性原理与Wagner-chottky模型考虑Mn的占据倾向。0 K时,Mn在Ni3Al中无明显占据倾向。有限温度内,随着温度的升高,在Ni3Al的富Al合金中,Mn由占据Ni亚晶格转变为占据Al亚晶格;在正常化学计量的合金中,Mn由无明显占据倾向转变为占据Al亚晶格;富Ni的合金中,Mn占据Al亚晶格。这一结论与实验结果相符。 由此可见,位置优先能只简单考虑了Mn直接替代Ni亚晶格及Ni亚晶格的单一缺陷情况,而转移能的判断更进一步考虑到反位缺陷与元素取代同时产生的情况。第一性原理结合Wagner-Schottky模型可以同时考虑温度及合金成分对Mn占位情况的影响。随着成分以及温度的变化,Mn的占据倾向也将发生相应的变化。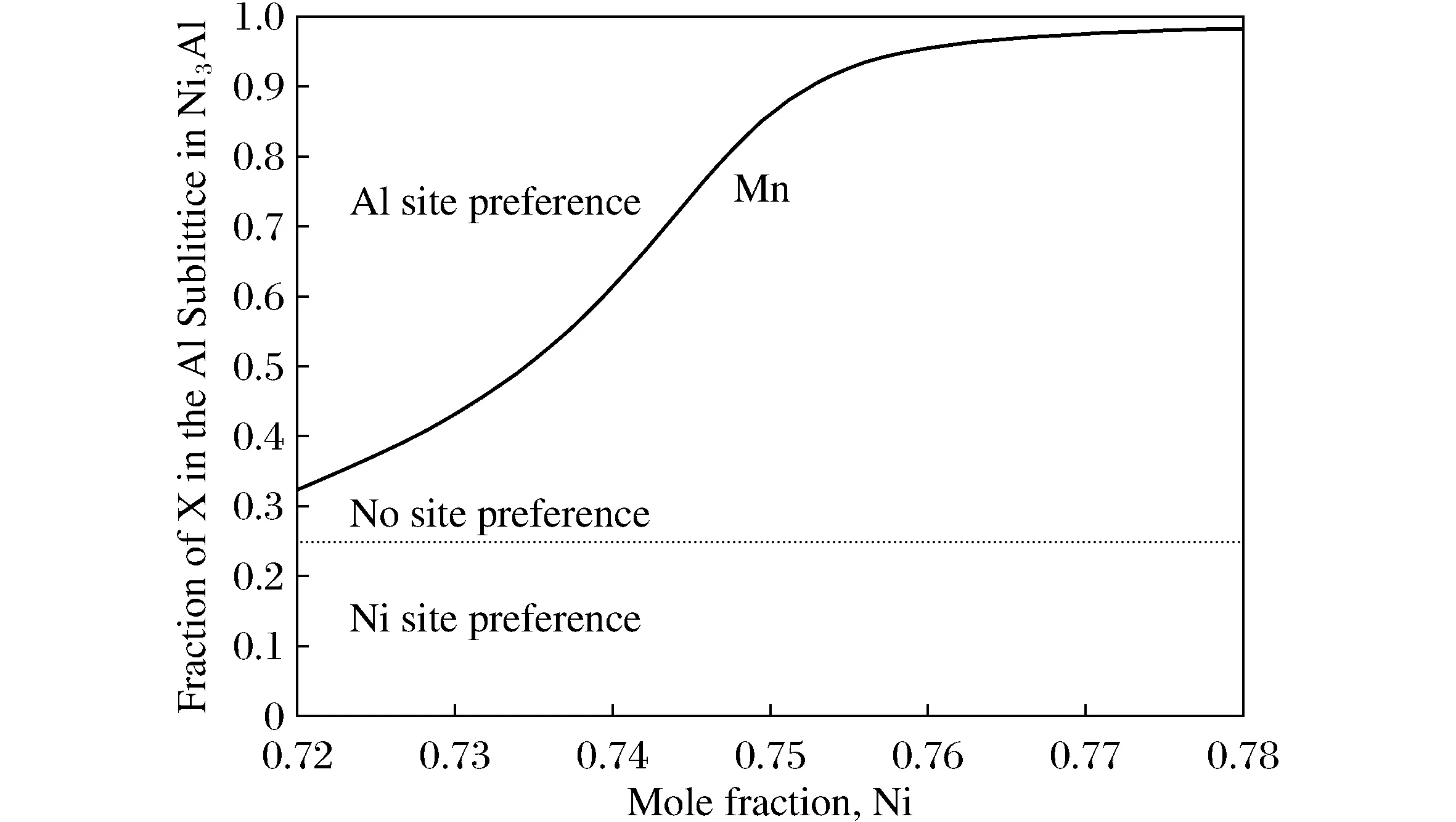
3 结 论
猜你喜欢
太原理工大学学报(2022年5期)2022-09-23
教育家(2022年19期)2022-05-13
当代作家(2021年11期)2021-12-17
成都信息工程大学学报(2021年2期)2021-07-22
科学(2020年4期)2020-11-26
华东师范大学学报(自然科学版)(2020年1期)2020-03-16
科学(2020年4期)2020-01-11
商周刊(2018年25期)2019-01-08
传媒评论(2018年5期)2018-07-09
中国卫生(2016年12期)2016-11-23
