
3,4-Secocycloartane Triterpenoids from the Cones of Pseudolarix amabilis
2021-03-12 02:17:06SiJiaXiaoBoLiZhengRuiHuangWenLinYuanJiYeHuiLiangLiXiKeXuYunHengShenWeiDongZhang
Si-Jia Xiao ·Bo Li ,2·Zheng-Rui Huang ·Wen-Lin Yuan ·Ji Ye ·Hui-Liang Li ·Xi-Ke Xu ·Yun-Heng Shen ·Wei-Dong Zhang ,2
Abstract Four new 3,4-secocycloartane triterpenoids, pseudolactones A—D (1— 4), were isolated from the ethanol extract of the cones of Pseudol arixamabilis.Their structures were established by extensive 1D- and 2D-NMR experiments.The cones of P.arixamabilis are enriched in the ring-expanded or cleaved cycloartane triterpenoids.This work provides new insight into cycloartane triterpenoids from the cones of P.arixamabilis.
Keywords Pseudol arixamabilis·Triterpenoids·3,4-Secocycloartane·Pseudolactones A—D
1 Introduction
Pseudolarix amabilisis a plant indigenous to the south-east of China [1].The root and trunk barks of it, known as ‘Tu Jin Pi’ in traditional Chinese medicine, have been used to treat skin diseases caused by fungal infection [2,3].Previous phytochemical studies on the root barks and seeds ofP.amabilisrevealed a variety of bioactive compounds with novel structures, with the main chemical constituents being pseudolaric acid analogous and triterpenoids [4— 9].Some of them, such as psedolaric acids A and B [10], have shown potent antimicrobial and cytotoxic activities.Peudolarolide B, a triterpene lactone, has shown potent cytotoxic activity [8].Novel nortriterpenoid lactone, pseudolarenone [11], as well as triterpenoid—diterpenoid dimers [12], have also been reported from the cones of this plant.In this paper, we describe the isolation and structure elucidation of four new 3,4-secocycloartane triterpenoids from the cones ofP.amabilis.
2 Results and Discussion
The dried cones ofP.amabilis, collected in Jiujiang, Jiangxi province, P.R.China, were extracted with 80% EtOH for three times at room temperature.The extract was separated by chromatography techniques to yield four new triterpenes, pseudolactones A—D (1— 4) (Fig.1).The structures of four new triterpenoids were determined by analysis of HRESIMS and NMR spectroscopic data.
Compound 1 corresponds to the molecular formula C32H50O6as established by the hydrogen adduct ion peak atm/z531.3678 [M + H] + (calcd.531.3680 for C32H51O6+) in HR-ESI—MS spectrum, indicative of 8 degrees of unsaturation.
The1H NMR spectrum (Table 1) of 1 showed four tertiary (δH1.00 (s), 1.17 (s), 1.18 (s), 1.08 (s)) and two secondary (δH0.86 (d,J= 6.4 Hz), 1.19 (overlap)) methyls.Moreover, an oxygenated proton signal was observed atδH4.05.In13C NMR spectrum (Table 2), there existed 32 carbon resonances, which were sorted into seven methyls, eleven methylenes, six methines (including one oxygenated methane atδC77.4), and eight quaternary carbons (including two ester carbonyls atδC174.6 and 179.7, an oxygenated quaternary carbon atδC76.1, and a ketal carbon atδC107.3), by DEPT NMR spectrum.The above evidences, combined with the characteristic methylene protons atδH0.51 (d,J= 4.4 Hz) and 0.70 (d,J= 4.4 Hz) as well as the carbon resonances atδC32.4 (t), 21.8 (s), and 27.6 (s), revealed a cyclopropyl motif.The diagnostic chemical shifts for two ester carbonyls atδC174.6 (C-3) and 179.7 (C-26), one oxygenated methine atδC77.4, and the ketal carbon atδC107.3, implied that compound 1 might be a 3,4-secocycloartane triterpenoid with a unique 16,23-epoxy-23,26-spirolactone side chain.
Comparison of the NMR spectroscopic data of compound 1 with those of known pseudolarolide C [8] indicated that they were structurally quite similar except that an additional ethoxy in compound 1 (δH1.19 (3H), 4.05 (2H);δC14.1, 60.2) replaced the methoxyl in pseudolarolide C, which was confi rmed by key 1 H- 1 H COSY correlations (Fig.2) of H2-1′/H3-2′ and HMBC correlation from H2-1′ (δH4.05) to carboxylic carbon C-3 (δC174.6 ppm).The structure of 1 was further evidenced by key 1 H- 1 H COSY correlations of H2-2/H2-1, H-5/H2-6/H2-7/H-8,H2-11/H2-12,H2-15/H-16/H-17/H-20 (H3-21)/H2-22 together with key HMBC correlations (Fig.2) from H2-2,H2-19,H2-6 to C-10 (δC27.6), from H2-11, H-15 to C-13 (δC43.5), from H2-12, H-16 to C-14 (δC47.3), from H2-22, H-16,H2-24 to C-23 (δC107.3), and H3-27 to C-26 (δC179.7).The relative confi gurations of 1 were elucidated to be identical with those of pseudolarolide C on the basis of the REOSY correlations (Fig.2) of H-19 with H-8, H-8 with CH 3 -18,CH3-18 with H-16, H-16 with H-20, H-17 with CH3-28 and CH3-21, H-22 with H-24, and H-24 with CH3-27.In cycloartane triterpenoids, the C-20 position of the 17-side chain is usuallyR-confi guration.From the biogenetic point of view,the C-20 position of 1 should beR-confi guration, too.Zhao et al.reported a serial of similar 16,23-epoxy-26(23)-olide-3,4-secocycloartan-3-oic acid esters, and determined their absolute confi gurations by single crystal X-ray diffraction [13].By comparing the Cotton eff ect (- 0.7 at 223 nm) of 1 in ECD spectrum with those known compounds [13], in combination with analysis of chemical shifts, the absolute confi gurations of C-23 and C-25 positions of 1 were proposed to beS- andR-confi guration, respectively.Thus, the structure of 1 was elucidated as (20R,23S,25R)-4-hydroxy-16,23-epoxy-26(23)-olide-3,4-secocycloartan-3-oic acid ethyl ester, and named pseudolactone A.
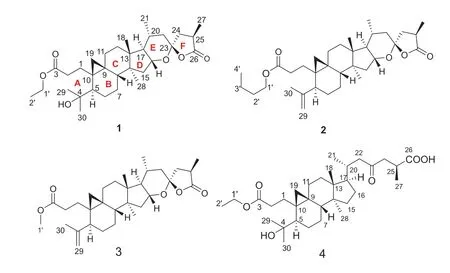
Fig.1 Chemical structures of 1— 4
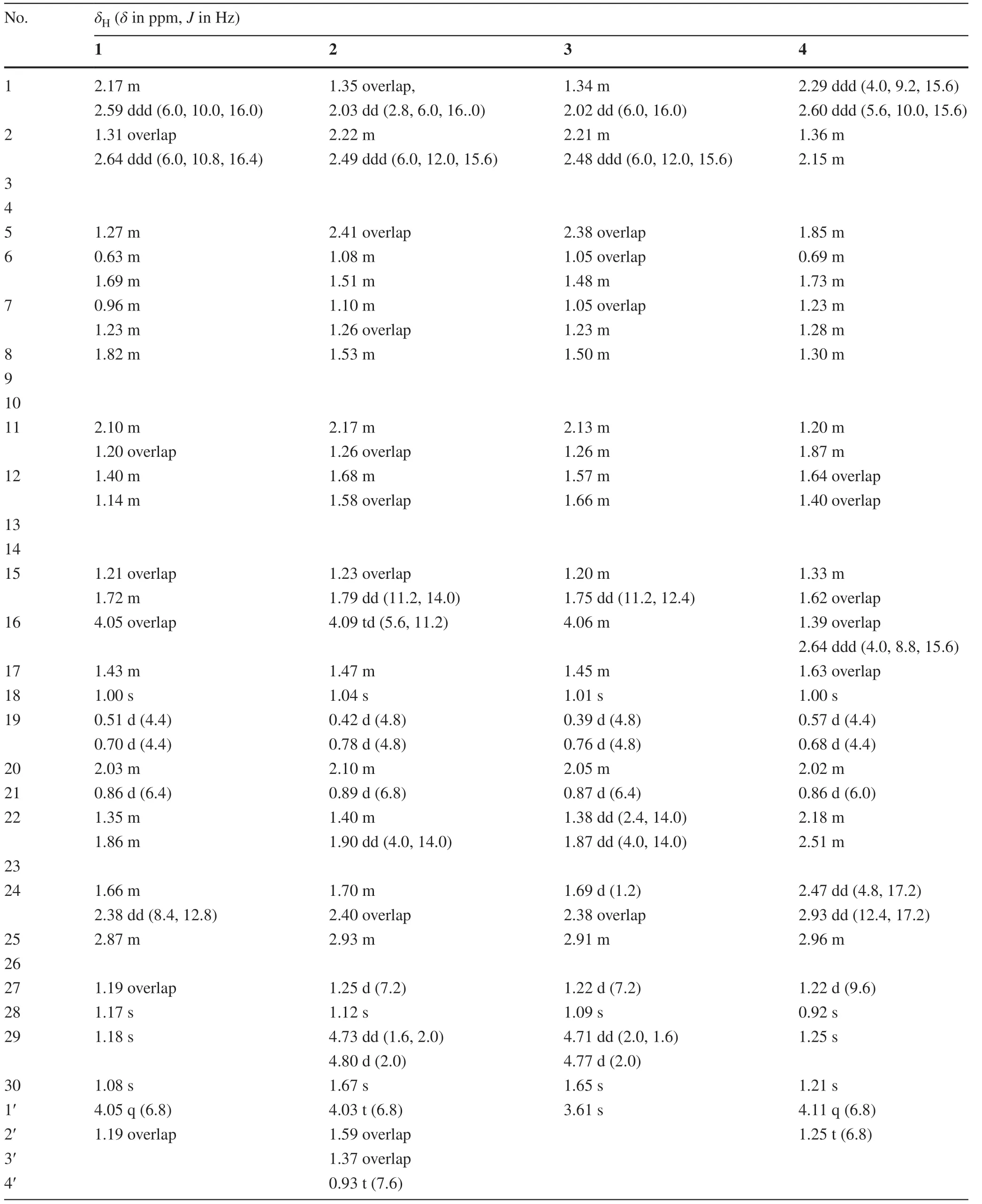
Table 1 1 H NMR (400 MHz) spectroscopic data for compounds 1— 4 in CDCl 3 (Jin Hz)

Table 2 13 C (100 MHz) NMR spectroscopic data of compounds 1— 4 in CDCl 3
Compound 2 was obtained as colorless oil.Its molecular formula was determined as C34H54O6, by HR-ESI—MS spectrum.The 1 H and 13 C NMR spectroscopic data (Tables 1 and 2) of 2 showed typical signals similar to those of 1, including two ester carbonyls (δC 174.0, 179.8), a ketal carbon atδC 107.3 (C-23), an oxygenated methine atδC 77.3 (C-16), a cyclopropyl atδH0.42 (d,J= 4.8 Hz), 0.78 (d,J= 4.8 Hz) andδC31.6 (t), one oxygen-bearing proton atδH4.09 (td,J= 5.6, 11.2 Hz) andδC77.3 (d), one oxygenated methylene atδH4.03 (t,J= 6.8 Hz) andδC64.2 (t), three singlet methyls atδH1.04 (s), 1.12 (s), and 1.67 (s), two doublet methyls atδH0.89 (d,J= 6.8 Hz) and 1.25 (d,J= 7.2 Hz), one triplet methyl atδH0.93 (t,J= 7.6 Hz).These information revealed that compound 2 possessed the similar 16,23-epoxy-26(23)-olide-3,4-secocycloartane skeleton to that of 1.The main differences between compounds 2 and 1 are that the resonance signals for one terminal double bond (δH 4.73 (dd,J= 1.6, 2.0 Hz), 4.80 (d,J= 2.0 Hz);δC 149.2 (s), 111.6 (t)), and for one butoxy (δH4.03, 1.59, 1.37, 0.93;δC64.2 (t), 30.6 (t), 19.1 (t), 13.7 (q)) in 2, took place of the signals for the oxygenated quaternary carbon atδC76.1 (s, C-4) and the methyl atδH1.17 (s) andδC25.8 (q), as well as for the ethoxy atδH4.05, 1.19 andδC60.2 (t), 14.1 (q).

Fig.2 Key 1 H- 1 H COSY, HMBC, and NOESY correlations of compound 1
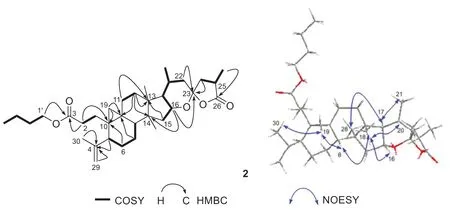
Fig.3 Key 1 H- 1 H COSY, HMBC, and NOESY correlations of compound 2
In 1 H- 1 H COSY spectrum (Fig.3), a butoxy was determined on the basis of the correlations of H2-1′/H2-2′/H2-3′/H3-4′.The butoxy was linked to C-3 through key HMBC cross peak (Fig.3) from H2-1′ (δH4.03) to the ester carbonyl atδC174.0.The terminal double bond was evidenced to be placed between C-4 and C-29 as shown by key HMBC correlations (Fig.3) from H-5 (δH2.41),H2-29 (δH4.73 and 4.80),CH3-30 (δH1.67) to C-4 (δC149.2).In REOSY spectrum of 2, the observation of key NOE correlations (Fig.3) of H-19 with CH3-30, H-8 (δH1.53), of CH3-18 (δH1.04) with H-8, H-16 (δH4.09) and H-20 (δH2.10), of H-17 (δH1.47) with CH3-28 (1.12) and CH3-21 (δH0.89), suggested that the relative confi guration of 2 was identical to that of compound 1.From structure, compound 2 could be considered as a dehydrated analogue of 1, implying that two compounds may share same confi guration.Also, compound 2 had a negative Cotton eff ect (- 1.5) at 201 nm.Consequently, the structure of 2 was identifi ed as (20R,23S,25R)- 16,23-epoxy-26(23)-olide-3,4-secocycloartan-4(29)-en-3-oic acidn-butyl ester, and named pseudolactone B.
The molecular formula of compound 3 was assigned as C31H46O5with nine degrees of unsaturation, based on the hydrogen adduct ion [M + H] + atm/z499.3414 (calcd.499.3418 for C31H47O5+ ).Its molecular weight is 42 Da less than that of 2.The 1 H and 13 C NMR spectra (Tables 1 and 2) were quite close to those of 2.The only difference was that a methoxyl atδH3.61 andδC51.5 was observed in 3, instead of those signals for the butoxy (δH4.03, 1.59, 1.39, 0.93;δC64.2 (t), 30.6 (t), 19.1 (t), 13.7 (q)) in 2.Key HMBC correlation from the methoxyl proton atδH3.61 to the ester carbonyl atδC174.3 further confi rmed the above deduction, and established the structure of 3 as (20R,23S,5R)-16,23-epoxy-26(23)-olide-3,4-secocycloartan-4(29)-en-3-oic acid methyl ester, and given the name pseudolactone C.
Compound 4 had a molecular formula C32H52O6with seven degrees of unsaturation, as evidenced by positive HRESI—MS atm/z555.3674 [M + Na] + .The 1 HNMR spectroscopic data (Table 1) of 4 exhibited four singlet methyls atδH1.00 (s), 0.92 (s), 1.25 (s), and 1.21 (s), two doublet methyls atδH0.86 (d,J= 6.0 Hz) and 1.22 (d,J= 9.6 Hz), one triplet methyl atδH1.25 (t,J= 6.8 Hz), and an oxygenated methylene atδH4.11 (2H, q,J= 6.8 Hz).In addition, a typical AB coupling system was observed atδH0.57 (1H, d,J= 4.4 Hz) and 0.68 (1H, d,J= 4.4 Hz).In 13 C NMR spectrum (Table 2), thirty-two carbon resonances were observed, including 7 methyls (δC 18.5, 19.2, 16.9, 19.5, 31.6, 26.2, 14.2), 12 methylenes (including one oxygenated methylene atδC60.3), 5 methines, and 8 quaternary carbons (including one ester carbonyl atδC174.8, one carboxyl atδC180.8, one ketone carbonyl atδC209.3, and one oxygenated quaternary carbon atδC76.3).Deducting three degrees of unsaturation accounted for one ketone carbonyl and two carboxylic carbon, the remaining four degrees of unsaturation were indicative of the tetracyclic ring system of 4.The NMR spectroscopic data (Tables 1 and 2) of 4 quite resemble those of known compound pseudolarnoid G ((25S)-4-hydroxy-3,4-seco-cycloartan-23-one-3,26-dioic acid methyl), previously reported from the seeds of the tilted plant [13].The main differences are that compound 4 had an ethoxy ester and one carboxyl functionalities, while pseudolarnoid G had two methyl ester functionalities.
The diagnostic chemical shifts atδC174.8 (ester carbonyl) andδC76.3 (s) were indicative of oxidative cleavage of ring A between C-3 and C-4, and formed an ethyl ester at C-3 and an oxygenated isopropyl moiety at C-4.The deduction was evidenced by key 1 H- 1 H COSY correlation (Fig.4) between H2-1′ (δH4.11) and H2-2′ (δH1.25), and between H2-1 and H2-2, together with HMBC correlations (Fig.4) of H2-1 and H2-1′ with C-3 (δC174.8), of CH3-29 (δH1.25),

Fig.4 Key 1 H- 1 H COSY, HMBC, and NOESY correlations of compound 4
CH 3 -30 (δH 1.21), H-5 (δH 1.85),H 2 -6 (δH 0.69, 1.73) with the oxygenated quaternary carbon atδC 76.3 (C-4).Also, the ketone carbonyl atδC209.3 was assigned to be C-23 on the basis of HMBC correlations from H2-22 and H2-24 to the ketone carbonyl.Key HMBC cross-peaks of H2-24 and CH3-27 (δH1.22 (d,J= 9.6 Hz) supported the attribution of C-26 carboxyl.In the ROESY spectrum, key NOE correlations (Fig.4) of H-20 with CH3-18, of H-8 (δH1.30) with CH3-18 and H-19, and of CH3-29 with H-19 indicated that H-8,CH3-18,CH2-19, H-20, and 4-hydroxyl isopropyl areβ-oriented, whereas H-17 and CH3-28 wereα-oriented based on the NOE cross-peak of H-17 with CH3-28.With regard to the absolute confi gurations of two chiral centers (C-20 and C-25) at C-17 side chain, Zhao et al.had ever determined the absolute confi gurations of C-20 and C-25 of similar compound pseudolarnoid G asRandS, respectively, by single crystal X-ray diffraction.The ECD spectrum of compound 4 showed a negative Cotton eff ect - 1.5 at 285 nm, in accordance with that of pseudolarnoid G (- 1.23 (283 nm)), revealed that compound 4 had same absolute confi guration as that of pseudolarnoid G.Therefore, the structure of 4 was identifi ed as (20R,25S)-4-hydroxy-23-oxo-3,4-secocycloartan-26-oic acid-3-ethyl ester, and named pseudolactone D.
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were measured with a Perkin—Elmer 341 polarimeter.IR spectra were recorded with a Bruker Vector-22 Spectrophotometer with KBr discs.NMR spectra were recorded with BrukerDRX-400spectrometer (400 MHz).The chemical shifts (δ) are given in ppm with TMS as internal standard and coupling constants (J) are given in Hz.MS spectra were recorded with aAgilent MSDTrap-XCT(for ESI) and Waters Micro-mass Q-TOF mass spectrometer (for HR-ESI—MS), inm/z.Column chromatographic separations were carried out by using silica gel (200—300 mesh;Marine Chemical Factory, Qingdao, P.R.China), Sephadex LH-20 (Pharmacia Fine Chemicals, Piscataway, NJ, USA) as packing material.TLC was carried out on precoated silica gel GF 254 plates (Yantai Chemical Industrials) and the TLC spots were viewed at 254 nm and visualized by using 10% sulfuric acid in alcohol containing 10 mg/mL vanillin.
3.2 Plant Material
The cones ofP.amabiliswere collected in Jiu Jiang, Jiangxi province, P.R.China, in October 2010, and authenticated by Prof.Han-Ming Zhang of Second Military Medical University.A voucher specimen (No.20101015) is deposited in School of Pharmacy, Second Military Medical University.
3.3 Extraction and Isolation
The air-dried cones (12.0 kg) ofP.amabiliswere ground into powder and extracted with 80% EtOH for four times at room temperature to give a crude extract, which was further partitioned with petroleum ether (60—90 °C) (PE),CHCl3, and EtOAc, successively.The CHCl3-soluable extract was subjected to a silica gel column chromatography (CC) eluting with a gradient PE/EtOAc (from 30:0 to 0:1) to obtain eight fractions 1—8.Fraction 2 (58 g) was chromatographed over RP-18 column eluting with MeOH/H2O (from 3:7 to 10:0) to aff ord five subfractions.Subfraction 2 was further chromatographed on a silica gel column (CH2Cl2/PE, from 0:20 to 1:0, and MeOH) and purifi ed by preparative TLC (cyclohexane/CH2Cl2/EtOAc, 20:1:1) to aff ord 2 (20 mg) and 3 (20 mg).Subfraction 4 was separated by repeated column chromatography on Sephadex LH-20 (CHCl3/MeOH, 1:1, and MeOH), and then purifi ed by preparative TLC (cyclohexane/CH2Cl2/EtOAc, 15:1:1) to yield 1 (10 mg).Fraction 7 (70 g) was separated by RP-18 CC (MeCN/H2O, from 2:8 to 10:0) to aff ord 6 subfractions.Subfraction 3 was further chromatographed on a silica gel column (CHCl 3 /MeOH from 50:1 to 0:1) and purifi ed by preparative TLC (PE/EtOAc/MeOH, 20:1:0.1) to aff ord 4 (8 mg).
3.3.1 Pseudolactone A (1)
Colorless oil, [α] 45.1 (c= 0.37,CH2Cl2).CD (c= 2.83 mmol/L,CH3CN, 20 °C) nm (Δε) 223 (-0.7).IR (KBr)νmax1731, 1778, 2964 cm -1 .For 1 H and 13 C NMR data (400 MHz,CDCl3), see Tables 1 and 2.ESI—MS:m/z553.5 [M + Na] + , 529.3 [M - H] - .HR-ESI—MS:m/z531.3678 [M + H] + (calcd C32H51O6+ , 531.3680).
3.3.2 Pseudolactone B (2)
Colorless oil, [α] 51.6 (c= 0.24,CH2Cl2).CD (c= 2.11 mmol/L,CH3CN, 20 °C) nm (Δε) 201 (- 1.5).IR (KBr)νmax1727, 1770, 2974 cm-1.For1H (400 MHz,CDCl3) and 13 C NMR data (100 MHz,CDCl3), see Tables 1 and 2.ESI—MS:m/z563.2 [M + Na]+, 539.1 [M - H] - .HRESIMS:m/z541.3902 [M + H]+(calcd C34H53O5+, 541.3888).
3.3.3 Pseudolactone C (3)
Colorless oil, [α] 51.1 (c= 0.23,CH2Cl2).CD (c= 2.61 mmol/L,CH3CN, 20 °C) nm (Δε) 201 (- 1.6).IR (KBr)νmax1737, 1778, 2964 cm-1.For1H (400 MHz,CDCl3) and13C NMR data (100 MHz,CDCl3), sees Tables 1 and 2.ESIMS:m/z521.2 [M + Na]+.HR-ESI—MS:m/z499.3414 [M + H] + (calcd C31H47O5+, 499.3418).
3.3.4 Pseudolactone D (4)
White amorphous powder, [α] 51.6 (c= 0.23,CH2Cl2).CD (c= 1.88 mmol/L,CH3CN, 20 °C) nm (Δε) 210 (- 1.9), 285 (- 1.5).IR (KBr)νmax1712, 1735, 2873, 2925, 2962, 3440 cm-1.For1H (400 MHz,CDCl3) and13C NMR data (100 MHz,CDCl3), see Tables 1 and 2.ESI—MS:m/z531.4 [M - H]-.HR-ESI—MSm/z555.3674 [M + Na]+(calcd C32H52O6Na+, 555.3656).
AcknowledgementsThe work was supported by NSFC (31870327, 81230090, 81520108030, 1302658), National Major Project of China (2018ZX09731016-005), The Key Research and Development Program of China (2017YFC1702002, 2017YFC1700200), Professor of Chang Jiang Scholars Program, Scientifi c Foundation of Shanghai China (17431902800, 16401901300), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (10DZ2251300).
Compliance with Ethical Standards
Conflict of interestThe authors declare that there are no conflicts of interest.
Open AccessThis article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creat iveco mmons.org/licen ses/by/4.0/.
 Natural Products and Bioprospecting2021年1期
Natural Products and Bioprospecting2021年1期
- Natural Products and Bioprospecting的其它文章
- Setosphlides A-D, New Isocoumarin Derivatives from the Entomogenous Fungus Setosphaeria rostrate LGWB-10
- Anti-microbial Eff ects In Vitro and In Vivo of Alstonia scholaris
- Four New Phloroglucinol-Terpene Adducts from the Leaves of Myrciaria cauliflora
- Asymmetric Total Synthesis of (+)-21- epi-Eburnamonine Via a Photocatalytic Radical Cascade Reaction
- Two New Quinazoline Derivatives from the Moss Endophytic Fungus Aspergillus sp.and Their Anti-inflammatory Activity
- Cytochalasins from Xylaria sp.CFL5, an Endophytic Fungus of Cephalotaxus fortunei