
Investigation of fluorescence resonance energy transfer ultrafast dynamics in electrostatically repulsed and attracted exciton–plasmon systems∗
2021-03-11 08:34HongYuTu屠宏宇JiChaoCheng程基超GenCaiPan潘根才LuHan韩露BinDuan段彬HaiYuWang王海宇QiDaiChen陈岐岱ShuPingXu徐抒平ZhenWenDai戴振文andLingYunPan潘凌云
Chinese Physics B 2021年2期
关键词:凌云
Hong-Yu Tu(屠宏宇), Ji-Chao Cheng(程基超), Gen-Cai Pan(潘根才), Lu Han(韩露),Bin Duan(段彬), Hai-Yu Wang(王海宇), Qi-Dai Chen(陈岐岱),Shu-Ping Xu(徐抒平), Zhen-Wen Dai(戴振文), and Ling-Yun Pan(潘凌云),†
1State Key Laboratory of Superhard Materials,College of Physics,Jilin University,Changchun 130012,China
2State Key Laboratory on Integrated Optoelectronics,College of Electronics,Science and Engineering,Jilin University,Changchun 130012,China
3State Key Laboratory of Supramolecular Structure and Materials,Jilin University,Changchun 130012,China
Keywords: fluorescence resonance energy transfer(FRET),quantum dots,excitons–plasmon composites
1. Introduction
Fluorescence resonance energy transfer(FRET)refers to fluorescence energy transfer from a donor fluorophore to an acceptor fluorophore. FRET can be combined with fluorescence quenching for optoelectronics, bio-labeling, the detection of heavy metals,etc.[1,2]Quantum dots(QDs)are replacing traditional fluorescent materials because of their benefits,which include tunable absorption peaks, large Stokes shifts,high quantum yields, and high compatibility.[3–5]FRET can be strongly enhanced for the detection of heavy metals in QDmetal nanoparticle (MNP) systems due to the local field effect generated by localized surface plasmons of excited metal NPs. Investigations of exciton (QD)–plasmon (MNP) interactions have progressed greatly in recent years. Hammer et al. studied CdSe-oligo(phenylene vinylene) (OPV) composite nanostructures and found blinking suppression in these QDs, which might be due to energy and charge transfer controlled by the ligands of QDs.[6]Mohammed et al.found that FRET occurs nonradiatively through a very short distance between the donor and acceptor.[7]Changwoo Nahm et al.also verified the optimum distance for the highest photoluminescence (PL) efficiency and the correlations between the calculated and measured quantum efficiencies in semiconductor/metal mixtures.[8]Excellent photostability can be obtained in an MUA–Au–CA–CdTe system for the detection of Pb2+ions, as reported by Wang and Guo.[9]Wang et al. investigated metallic microdisk-size dependence of QD’s spontaneous emission rate and micro-antenna directional emission effect for hybrid structures based on a particular single QD emission.[10]Research in this field has included investigations of fluorescence enhancement properties and the detection of mixed states,among other topics.[11–18]
The combination of QDs and MNPs shows various possibilities of adjusting fluorescent properties in nanoscale. MNPs with localized surface plasmon modes are an alternative option for enhancing the radiative recombination rate of a light emitter to increase the internal quantum efficiency of the system,the special characteristics of MNPs do not always play a positive role in other reports.[19–23]Most reported results are based on electrostatic attraction systems,which are similar to QDs and MNPs. However, QDs and MNPs other than those that are electrostatically attracted are involved in biomedical systems. When a target needs to be labeled and cured simultaneously, the labeling particle (QD) and curing particle(MNP) must have the same surface properties to attach to the target. Interactions between various nanomaterials thus need to be understood. Studying FRET-based luminescence quenching in an electrostatically repulsive system is therefore important since fluorescence detection is commonly used in biomedical treatments, and fluorescent dyes have been used to analyze the functions of internal granules, proteins, membranes, and nuclei.[24]Exciton and plasmon interactions can be analyzed kinetically by using time-resolved spectroscopy,which is helpful to understanding interactions between components and has been applied in fluorophore-labeled protein systems.[25–29]
In this article, exciton–plasmon interactions are investigated in a system with electrostatic repulsion; this system is composed of negatively charged thioglycolic acid (TGA)-CdTe QDs and negatively charged sodium citrate (NAC)-Au NPs. Kinetic processes are observed in the QD-MNP system by both steady-state and time-resolved spectroscopy. We found that FRET between QDs and metal NPs easily occurs under strong coupling conditions, while FRET among QDs is dominant with weak coupling. These two kinds of FRET cannot be distinguished well by steady-state spectra. Timeresolved spectroscopy can show the difference via the decay rates and amplitudes of each time component.
2. Experiments
2.1. Preparation of materials
Au NPs are prepared using the following compounds:sodium citrate (Na3C6H5O7, Tianjin Chemical Reagent),HAuCl4powder(99.8%,high-purity,Aldrich Company),and ultrapure water (electrical resistivity 18.2 MΩ/cm, Milli-Q).First 100 ml of 0.01% HAuCl4aqueous solution is heated to boiling. Then, 1.0 ml of a 1% Na-citrate aqueous solution is added to the above solution during stirring. The color changes from yellow to gray and then becomes black,soon turning red afterward. The whole process occurs in 2 min–3 min. The solution is then boiled for 15 min. After the solution is cooled to room temperature, distilled water is added to recover the original volume of the solution. Positively charged Au particles are prepared with hexadecyltrimethyl ammonium bromide(C16H33(CH3)3NBr,CTAB)as surface ligand instead of sodium citrate.[30]The synthesis of QDs has been described in Ref. [31]. The composites are prepared by mixing each QD with Au in a series of molar ratios and are named Au-QD1,Au-QD2(repulsion mode)and Au+QD1,Au+QD2(attractive mode),referring to each QD size.
2.2. Materials analysis
The sizes of Au NPs are observed by using a Hitachi H-800 IV SEM (200 kV). UV-visible absorption spectra of Au QDs are measured by a Shimadzu UV-3101PC.Luminescence spectra of QDs and composites are recorded by using a SHIMADZU RF-5301PC.
2.3. Materials analysis
Nanosecond fluorescence lifetime experiments are performed by a time-correlated single-photon counting(TCSPC)system with a right-angle sample geometry. A 405-nm picosecond diode laser (Edinburgh Instruments EPL375, repetition rate 2 MHz) is used to excite the samples. The fluorescence is collected by a photomultiplier tube (Hamamatsu H5783p)connected to a TCSPC board(Becker&Hickel SPC-130). The time constant of the instrument response function(IRF)is approximately 500 ps.
3. Results and discussion
3.1. Electrostatic repulsion
We choose two sizes of QDs for this investigation. The aim is to find the effect of resonance on energy transfer. Figure 1 shows the UV-visible absorption of Au NPs and the luminescence spectra of QDs. The resonance absorption peak of the NPs is located at 518 nm. The peak luminescence of the QDs is found at 525 nm(QD1)and 532 nm(QD2). According to these spectra,exciton–plasmon resonance can occur in QDs of both sizes. For comparison, luminescence quenching experiments are carried out with different exciton–plasmon composite ratios. The mixing ratio (Au:QD) is 1:5, 1:3, 1:1, 3:1,and 5:1.[32,33]And the luminescence quenching spectra of the composites are shown in Fig.2.
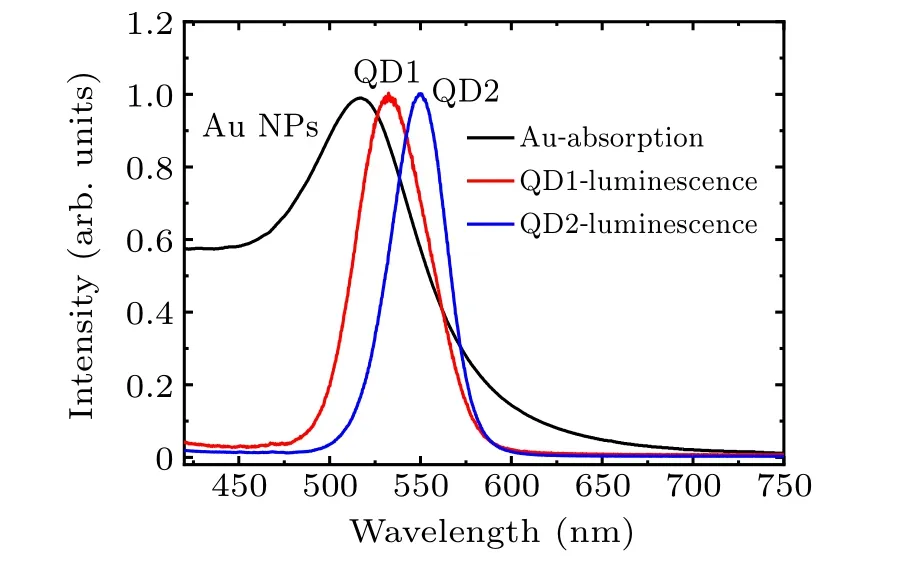
Fig.1. Normalized UV-Vis absorption spectra of Au NPs(black solid line)and luminescence spectra of QDs(red QD1/blue QD2).
The band-edge absorption of QDs becomes broader with increasing Au particle concentrations for each composite. For the case with near resonance (Au-QD1), the amplitude of spectral broadening is slight for Au:QD1=1:5, 1:3, 1:1,and 3:1(FWHM=38 nm)and dramatic for Au:QD1=5:1(FWHM=52 nm, inset of Fig.2(a)). The shift of the luminescence peak (∆peak) is 30 nm. A gentler trend of spectral broadening appears in the Au-QD2 composite as does a dramatic increase in spectral width(inset of Fig.2(b)).
The luminescence intensity of Au-QD1 does not vanish rapidly with a spectral redshift (Fig.2(a)). The luminescence disappears incrementally with increasing Au NPs concentrations. Meanwhile,dramatic quenching accompanies a spectral redshift in Au-QD2 at mixing ratios greater than 1:1 (Fig.2(b)). The luminescence of Au-QD2 is quenched more dramatically than that of Au-QD1 with the same mixing ratio.[34]This result is caused by the strong resonance between the Au NPs and QD1. FRET easily occurs in a system with strong resonance (Au-QD1) when the distances between Au NPs and QDs are the same(the same mixing ratio). In the system with weak resonance(Au-QD2),FRET occurs only when the distance between Au and QDs is close enough. For system with strong resonance, no clear redshift occurs with quenching.In contrast,a redshift suddenly appears at mixing ratios up to 1:1 in the system with weak resonance. The redshift could be due to either local-field-induced mixing of states in QDs or steric hindrance-induced aggregation of QDs with increasing of Au particle concentrations. Decreases in the distances between QDs can also induce FRET, but this FRET causes a redshift as a spectral signature. This process should be slower than local-field-induced FRET. The dynamics results give a reasonable explanation for this phenomenon.
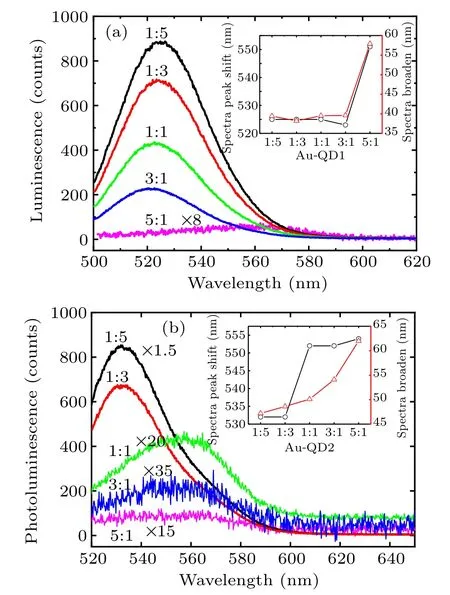
Fig.2. Photoluminescence spectra of the Au-QD composites Au:QD1 (a)and Au:QD2(b)with different mixing ratios: 1:5 black,1:3 red,1:1 green,3:1 blue,5:1 pink.
The dynamics of the process of luminescence quenching is observed by TCSPC. The results of the experiments are listed in Fig.3. The fitting parameters are shown in Table 1.Both the lifetimes and amplitudes of Au:QD1 slightly decrease with increasing quantities of Au, the lifetimes range from 4.0 ns (16%), 8.6 ns (32%), and 25.9 ns (52%) for Au:QD1=1:5 , but the lifetimes range for Au:QD1=3:1 is 1.9 ns (22%), 8.3 ns (36%), and 25.6 ns (42%) (Fig.3(a)& Table 1). As the Au:QD1 value reaches 5:1, the lifetimes and their amplitudes suddenly decrease,with values of0.7 (65%), 3.6 (28%), and 21.4 ns (7%). Ninety percent of the luminescence is quenched within 4 ns, and a large luminescence redshift of 80 nm is observed. This quenching is much faster than that of aggregation-induced luminescence quenching.[35,36]The quenching is thus induced by energy transfer between Au and QD1 particles. The rate of this process is limited by the distance between particles. Since both Au and QD1 are negatively charged, the barrier for energy transfer is higher when the distance between them is large,for example, when Au:QD1=1:5, 1:3, 1:1, or 3:1. The distance between Au and QD1 particles decreases with the space compression due to increased Au particle steric hindrance. Localfield-induced FRET can occur and induce dramatic change in dynamics at Au:QD1=5:1 (Fig.3(a)). This result is consistent with the spectrum broadening induced by the local-fieldinduced hybrid states (FWHM=58 nm, inset of Fig.2(a));the width of this spectrum is much larger than that of an aggregation-induced spectrum (FWHM=39 nm).[35,36]The dynamic change as the mixing ratio changes is much gentler in Au-QD2(Fig.3(b)).The decay rates of the fast and medium components do not change much between the Au:QD2=1:5,1:3,1:1,and 3:1 samples,which exhibit lifetimes of ~0.6 ns and ~4.5 ns. The amplitude of the fast component increases from 59%to 81%with the disappearance of the slow component at ~20 ns.The fast component in the Au:QD2=5:1 sample lives for 0.3 ps,which is close to the resolution of this TCSPC setup. The average lifetime is calculated to be less than 1 ns,and the initially photoexcited hot carriers thermalize and redistribute primarily through electron–electron(e–e)scattering within tens of femtoseconds (fs). Since the decay rate of the fast component is on the scale of local-field-enhanced FRET, the luminescence quenching in weakly resonant Au-QD2 may be due to FRET between Au and QDs.[30,31]These results are consistent with the slight broadening of the luminescence spectra of Au:QD2=1:1, 3:1, and 5:1 systems(FWHM=50 nm–62 nm), which occurs at lower Au ratios than in the Au-QD1 system and suggests that local-fieldinduced FRET occurs (inset of Fig.2(b)). The difference in Au-QD1 and Au-QD2 results from the easier aggregation of QD1 than QD2 as a result of the higher surface energy and less electrostatic repulsion of QD1. In summary,aggregationinduced FRET among QDs at the same Au:QD ratio is the main contributor to the quenching process in the strongly resonant/small composite (Au-QD1).[37]In contrast, local-fieldenhanced FRET between Au and QDs is the main contributor to luminescence quenching in the weakly resonant/large composite(Au-QD2).

Table 1. Fitting parameters of the luminescence lifetime of Au-QD composites.

Fig.3. The luminescence decay of the Au-QD composites Au:QD1(a)and Au:QD2 (b) with different mixing ratios: 1:5 black, 1:3 red, 1:1 green, 3:1 blue,5:1 pink,IRF(system reference scattering)circle+solid gray line.
3.2. Electrostatic attraction
To compare electrostatic effects, we repeated the above experiments in Au+QD composites in which the Au is positively charged. Figures 4(a) and 4(b) show that absorption spectra slightly redshift as well as broaden at the band-edge for both composites. Luminescence redshifts and becomes asymmetrical as the mixing ratio increases to Au:QD=1:1. The Au+QD1 composite luminescence extends further at the bandedge (∆peak=24 nm) than the Au+QD2 composite luminescence (∆peak=18 nm). The luminescence of Au+QD2 redshifts more clearly at mixing ratios up to 1:1(∆peak=28 nm)than that of Au+QD1 (∆peak= 6 nm). The larger redshift of luminescence in Au+QD2 is caused by a strong electrostatic effect, which is the same factor as that in the Au-QD system,as saying, either local-field-induced mixing of states in QDs or steric hindrance-induced aggregation of QDs. For mixing ratio Au:QD2=5:1,∆peak=21 nm for Au+QD1,and∆peak=32 nm for Au+QD2. The quenching is stronger in Au+QD2 than Au+QD1 for the same mixing ratio. At a particular distance,FRET occurs more easily with the strong resonance in Au+QD1. With the weak resonance in Au+QD2,FRET does not occur until the distance between Au and QDs is short enough.Strongly and weakly resonant systems show obvious redshifts in luminescence in the Au+QD systems. This result can be due to the enhanced electrostatics between negative QDs and positive Au, which reduces the distance between particles. The local-field effect is thus stronger in the Au+QD systems,and hybrid states are more easily generated,which leads to the non-Gaussian shapes of their luminescence spectra.[38–40]

Fig.4. The photoluminescence of the Au+QD composites Au:QD1(a) and Au:QD2 (b) with different mixing ratios: 1:5 black, 1:3 red, 1:1 green, 3:1 blue,5:1 pink.
Considering the slight broadening of spectra and the fast component(<50%)with a lifetime>1 ns in dynamics,both Au+QD1 and Au+QD2 show aggregation-induced quenching at the low Au:QD ratios of 1:5 and 1:3. When Au:QD is 1:1,local-field-induced quenching appears in the large QD system(Au+QD2) (0.5 ns (87%), Fig.5 and Table 2). This result is consistent with spectral changes in Figs. 4(a) and 4(b), in which the spectra are broadened when average lifetimes <3 ns and fast components that decay in less than 1 ns appear. The situation is similar to the Au-QD system. At low Au:QD ratios, aggregation-induced FRET leads to lifetimes > 10 ns and the slow component accounting for over 30% of the decay, accompanied by small changes in the spectra (Table 2).At high Au:QD ratios,local-field-induced FRET leads to lifetimes <3 ns and a fast component that accounts for over 60%of the decay, accompanied by greater broadening of spectra.The difference results in Au-QD2 having a faster lifetime than Au+QD2 at Au:QD=1:5 and 1:3;these lifetimes are 3 ns–5 ns in Au-QD2 and 11 ns–15 ns in Au+QD2. This result may be due to QD aggregation on the Au particles’surface in the electrostatically attracted system,which is the main contributor to FRET in the weakly resonant Au+QD system(Au+QD2).

Fig.5. The luminescence decay of the Au+QD composites Au:QD1(a)and Au:QD2(b)with different mixing ratios: 1:5 black,1:3 red,1:1 green,3:1 blue,5:1 pink,IRF(system reference scattering)circle+solid gray line.

Table 2. Fitting parameters of the luminescence lifetime of Au+QD composites.
4. Conclusion
FRET between excitons (QDs) and plasmons (Au NPs)was observed in both electrostatically repulsed and attracted systems. Steady-state spectroscopy shows that luminescence quenching occurs in both systems with increasing Au quantities. However, time-resolved spectroscopy indicates that aggregation-induced FRET between QDs is a priority compared to the FRET between Au and QDs at low Au:QD ratios in both electrostatically attracted and repulsed systems,which show lifetimes of tens of ns and an ns fast component. Localfield-induced FRET is observed when the steric hindrance of plasmons is improved at high Au:QD ratios, resulting in ns lifetimes and fast components decaying at hundreds of ps. For different sized QDs, aggregation-induced FRET is prominent in small QDs and local-field-induced FRET is prominent in large QDs.The above process is difficult to identify using only steady-state spectroscopy. Aggregation-induced FRET cannot be ignored in any environment regardless of the electrostatic conditions. Local-field-induced FRET appears when Au concentrations reach a certain threshold.The nature of FRET cannot be determined by steady-state spectroscopy only. Under complicated conditions, time-resolved spectroscopy must be employed too.
猜你喜欢
做人与处世(2022年2期)2022-05-26
应用数学(2020年3期)2020-07-28
中学时代(2019年12期)2020-01-02
兵器知识(2019年1期)2019-01-23
陶山(2018年4期)2018-12-28
航空世界(2018年12期)2018-07-16
北方音乐(2018年15期)2018-05-14
北方音乐(2018年4期)2018-05-14

- Chinese Physics B的其它文章
- Novel traveling wave solutions and stability analysis of perturbed Kaup–Newell Schr¨odinger dynamical model and its applications∗
- A local refinement purely meshless scheme for time fractional nonlinear Schr¨odinger equation in irregular geometry region∗
- Coherent-driving-assisted quantum speedup in Markovian channels∗
- Quantifying entanglement in terms of an operational way∗
- Tunable ponderomotive squeezing in an optomechanical system with two coupled resonators∗
- State transfer on two-fold Cayley trees via quantum walks∗