
水溶性淀粉在蒙脱土和高岭土上的吸附保护
2021-03-09 17:44尹梦楠
昆明理工大学学报(自然科学版) 2021年1期
张 璐,石 林,尹梦楠,张 凰
(1.昆明理工大学 环境科学与工程学院,云南昆明650500;2.昆明理工大学农业与食品学院,云南昆明650500)
0 引言
土壤作为最大的碳库,每年有机碳流失量为75~100 Pg C[1-2],比人类活动释放碳的速率高一个数量级,因此,研究者对土壤有机质的稳定性进行了广泛的关注,发现除受自身物理、化学、结构特征的影响外,矿物的吸附保护也是极为重要的稳定机制.例如:金属氧化物通过配体交换作用优先吸附保护不饱度低、含氧量高的芳香组分[3-4];黏土矿物则通过疏水作用优先吸附保护饱和度高、含氧量低的脂类物质[5-6].以往这些研究普遍关注的是以腐殖质为代表的土壤有机质稳定组分,需要指出的是,在土壤有机质各主要组分中,根际分泌物和生物质中可溶性组分如碳水化合物、蛋白质、有机酸等的稳定性差,极易被降解和氧化,半衰期仅为数周[7],对土壤碳排放贡献巨大,是影响土壤碳行为的关键土壤组分(非腐殖质活性组分)[8].土壤非腐殖质活性组分输入量巨大,占土壤新输入有机质的一半以上[9].而且由于其较高的降解流失率,对碳排放的贡献远远大于土壤有机质稳定组分的贡献[8].然而其溶解性大、易于被降解和氧化的性质,致使其被外界所忽略,事实上,土壤非腐殖质活性组分的稳定是土壤有机质稳定性提高的核心环节.然而,到目前为止,还没有研究系统阐述土壤非腐殖质活性组分受矿物吸附保护的特征和机制.
此外,研究者普遍关注于矿物的吸附保护对腐殖质的稳定机制.对于土壤非腐殖质活性组分而言,由于其具有较为简单的结构、较小的分子量和较大的溶解度,可能更有利于其在土壤矿物上吸附保护.其次,由于土壤非腐殖质活性组分自身物理、化学、结构的不稳定性,矿物的吸附保护对其稳定性的提高可能贡献更大.然而,到目前为止,还没有研究系统阐述土壤非腐殖质活性组分受矿物吸附保护的特征和机制.由于矿物种类多样,晶型结构复杂,不同矿物,甚至晶型结构不同的同种矿物,对土壤非腐殖质活性组分的吸附保护特征可能也完全不同.例如:1∶1型黏土矿物(如高岭石)层间为氢键紧密联结,层间距极小且不可变,可能仅对溶解度较大的小分子有机酸等组分提供保护;而蒙脱土等2∶1型黏土矿物的层间距可以随含水量的增大而增大[10],对溶解度较大的大分子蛋白质等组分也可能提供保护.然而,对于矿物类型特别是晶型结构如何影响其对土壤非腐殖质活性组分吸附保护作用的研究还鲜有报道.
基于以上论述,本研究选择输入量大且易降解的水溶性淀粉作为代表性的土壤非腐殖质活性组分,选取晶型结构不同的高岭土和蒙脱土,通过批量等温吸附实验,探究两种矿物对水溶性淀粉的吸附特征和机制.通过批量高锰酸钾氧化实验,探究矿物吸附前后,水溶性淀粉的抗氧化性,用以评估矿物吸附保护的贡献,为探索土壤非腐殖质活性组分在土壤固碳中的关键作用提供理论依据.
1 材料及方法
1.1 实验材料
实验所用水溶性淀粉,购于上海阿拉丁生化科技股份有限公司,纯度为分析纯;高岭土(Kao,粒径为11μm),购于上海阿拉丁生化科技股份有限公司;蒙脱土K10(Mon)购于阿法埃莎化学有限公司;盐酸、氢氧化钠均购置于四川西陇化工有限公司,纯度为分析纯;高锰酸钾购置于广东汕头化工厂,纯度为分析纯;实验所用超纯水由Milli-Q Reference超纯净水机制备.
1.2 蒙脱土和高岭土的性质特征
通过BET比表面积测定仪(JW-BK132F,中国)对蒙脱土和高岭土的比表面积进行表征分析.通过湿式比表面分析仪(Xigo Nanotools Acorn Area,美国)对矿物的湿式表面进行分析.矿物100 mg,水土比为200∶1(质量比),调节pH至6.0,测定矿物悬浮液的H1弛豫时间(T).弛豫率(Rsp),即弛豫时间(T)的倒数,是吸附水和游离水弛豫率的加权平均数,可通过以下公式推导得出.当矿物浓度为C(mg/mL)时,吸附水占系统中所有水的比例为:

整理得:

式中:ɑ为吸附水占系统中所有水的比例,c为每一个质子(氢原子核)在束缚态的时间分数,C为颗粒浓度(mg/mL),Tbound和Tfree分别为自由和束缚溶剂的弛豫速率常数,Rsp为弛豫率,T为弛豫时间.颗粒浓度相同时,弛豫率Rsp可反映颗粒表面吸附水的量多少,即可有效反映颗粒吸附水分子的能力大小[11],进而体现颗粒表面的亲水性及极性有机质亲和性.
通过傅里叶变换红外光谱仪(Varian640-IR,美国)表征蒙脱土和高岭土的化学官能团类型.样品制备采用KBr压片法,KBr与矿物比例为200∶1,样品扫描波数范围为400~4 000 cm-1,分辨率为2 cm-1,图谱由扫描50次累加而后自动基线校正和自动平滑处理.
1.3 蒙脱土和高岭土对可溶性淀粉的吸附实验
按照预实验确定的固液比1∶200(质量比),称取100 mg矿物(Kao/Mon)于40 mL棕色瓶内,加入10 mL去离子水,用0.10 mol/L HCl或0.10 mol/L NaOH溶液调节溶液pH至(6.0±0.1);分别配制浓度为20~200 mg/L的水溶性淀粉溶液,调节溶液pH至(6.0±0.1),取10 mL加入至上述棕色瓶中.根据预实验吸附动力学平衡时间,全部样品于暗处25℃下以120 r/min速度振荡24 h.悬浊液以3000 r/min的速度离心15 min,取10 mL的上清液,用0.45μm水相针式过滤器过滤,通过高效液相色谱(Agilent1260)对过滤液中的水溶性淀粉进行定量分析.具体方法为:糖柱(安捷伦,Hi-Plex H);示差折光检测器;流动相pH=6.0的HCl溶液,流速为1.5 mL/min,进样量为20μL,柱温箱温度为30℃.本实验设置2个平行处理.
根据方程(4)计算蒙脱土和高岭土对水溶性淀粉的吸附量:

式中:Qe为矿物对水溶性淀粉的吸附量,mg/g;C0和Ce分别为水溶性淀粉初始和吸附平衡时的浓度,mg/L;V为溶液体积,L;m为矿物质量,g.
吸附等温线以Langmuir和Freundlich模型拟合,公式如下:
Langmuir模型:

Freundlich模型:

式中:Qe和Qm分别为固体平衡吸附量和最大吸附量,mg/g;Ce为液相平衡浓度,mg/L;KL为Langmuir模型吸附系数,L/mg;KF为Freundlich模型吸附系数,(mg/g)·(mg/L)n;n为Freundlich常数.
1.4 高锰酸钾氧化降解动力学实验
称取1 g矿物(Kao/Mon)于250 mL棕色瓶内,加入100 mL去离子水,用0.10 mol/L HCl或0.10 mol/L NaOH溶液调节溶液pH至(6.0±0.1);配置浓度为200 mg/L的水溶性淀粉溶液,调节溶液pH至(6.0±0.1),取100 mL加入至上述棕色瓶中;同时设置无矿(None)的空白组作对照.于25°C下振荡24 h后,加入1 mL pH=6.0、浓度为120 mmol/L的高锰酸钾溶液,振荡后分别在1、2、5、10、15、20、30、60、120、180 min取样,过0.45μm水相针式过滤器.实验采用紫外可见分光光度计(UV-2600,日本岛津)检测高锰酸钾的吸光度.本实验设置2个平行处理.
2 结果与讨论
2.1 蒙脱土和高岭土性质特征分析
以水溶性淀粉为代表的有机碳在矿物表面的吸附与矿物的表面物理特征和化学特征密切相关.借助BET和Xigo对两种矿物进行表征,结果如表1所示.BET测定结果表明,蒙脱土的比表面积(200.76 m2/g)远远大于高岭土的比表面积(6.10 m2/g),但是高岭土具有较大的孔径,与Septian等人研究相符[12].基于N2吸附的BET法测量的是干燥矿物的比表面积,无法直观体现矿物在溶液中可被利用的表面[13].Xigo湿式比表面分析仪基于核磁共振原理测定矿物溶液的弛豫时间(T),用以描述溶剂中的颗粒表面的性质.矿物颗粒浓度相同时,Rsp(Rsp=Tfree/T-1)反映了颗粒吸附水分子的能力和颗粒表面的亲水性大小[8].蒙脱土Rsp大于高岭土,表明蒙脱土颗粒表面吸附水分子的能力强于高岭土,蒙脱土颗粒表面的亲水性更大[11].

表1 高岭土和蒙脱土的BET和Xigo数据Tab.1 Bet and Xigo data of kaolin and montmorillonite
借助傅里叶红外光谱进一步印证蒙脱土的亲水性.如图1所示,高岭土和蒙脱土的表面官能团种类大致相同,高岭土和蒙脱土的Si-O-Si键在468 cm-1处弯曲振动;Si-O-Al键分别在560、731 cm-1和530、799 cm-1处发生弯曲振动;Si-O键伸缩振动则分别在1 100 cm-1和1 051 cm-1处出现[14].而关于吸附水的弯曲、伸缩振动则分别发生于1 640、3 445 cm-1和1 640、3 445 cm-1[14].需要强调的是,蒙脱土还在3 620 cm-1处产生内表面的-OH键振动[14],表明蒙脱土表面有更多的水分子存在,与Xigo湿式比表面分析仪测定结果相吻合.高岭土和蒙脱土的波数差异主要与其矿物类型有关.高岭土属于1∶1型层状硅酸盐类矿物,层间为氢键紧密联结,层间没有水分子和阳离子,层间距固定.矿物颗粒较大,呈六角形片状,比表面小,以外表面为主,阳离子交换量很低[15].蒙脱土则是2∶1型层状硅酸盐粘土矿物,晶层间通过静电和van der Waals键结合.层间能吸水膨胀,有较强的膨胀性,矿物颗粒较小,有很大的比表面积,且以内表面为主[15].

图1 高岭土和蒙脱土的傅立叶红外光谱图Fig.1 FTIR spectra of kaolin and montmorillonite
综合来看,两种矿物的晶形结构决定了蒙脱土的吸附以内表面为主,高岭土的吸附以外表面为主.此外,蒙脱土颗粒表面吸附水分子的能力强于高岭土,水分子优先通过氢键吸附于蒙脱土表面,形成一层水化膜[16].
2.2 蒙脱土和高岭土对水溶性淀粉的吸附特性
高岭土和蒙脱土吸附水溶性淀粉的等温线通过Langmuir和Freundlich模型拟合如图2所示,两种模型均显出较好的拟合效果,相应的拟合参数列于表2.水溶性淀粉在高岭土上的吸附远远大于在蒙脱土上的吸附.从Langmuir拟合结果来看,水溶性淀粉-高岭土体系的吸附系数KL(1.51×10-2L/mg)约为水溶性淀粉-蒙脱土体系(0.41×10-2L/mg)的4倍.然而,从饱和吸附量来看,蒙脱土的最大吸附值Qm(5.58 mg/g)大于高岭土(3.98 mg/g).Freundlich拟合结果也显示,水溶性淀粉-高岭土体系的平衡系数KF(0.15(mg/g)·(mg/L)n)约是水溶性淀粉-蒙脱土体系(0.034(mg/g)·(mg/L)n)的4倍,且均为非线性吸附(n<1).
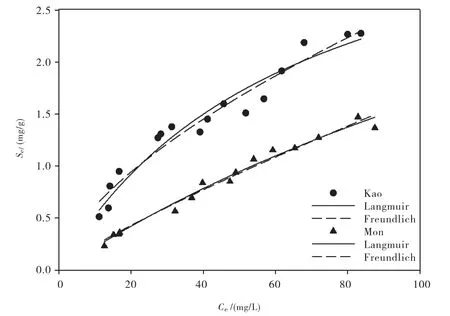
图2 高岭土和蒙脱土对水溶性淀粉的吸附等温线Fig.2 Adsorption isotherms of water soluble starch on kaolin and montmorillonite

表2 高岭土和蒙脱土对水溶性淀粉的吸附等温线拟合参数Tab.2 Fitting parameters of adsorption isotherms of water soluble starch by kaolin and montmorillonite
水溶性淀粉主要是由支链淀粉构成,基本结构单元是α-D-吡喃葡萄糖,结构排列不规则,分子量可达几百万,结晶度差,可溶于冷水,粘度大[17].水溶性淀粉的单体物质葡萄糖是一种多羟基的有机质,可以通过氢键作用吸附到矿物表面[18].此外,憎水性作用也是水溶性淀粉与黏土矿物相互作用的重要机制之一[19].水溶性淀粉占据黏土矿物高岭土和蒙脱土表面的吸附位置后,在矿物表面形成有机分子层,为继续吸附水溶性淀粉创造了潜在的吸附点位[20],可能表现出多层吸附.水溶性淀粉浓度越高,“进入”矿物表面较规则的层状“矿物-有机质”复合体中更容易,表现出更大的吸附量[21],类似于一种有机质“叠层”模型——有机质在矿物表面形成了类似洋葱的层状结构[22].一般而言,黏土矿物对有机质的吸附主要与本身的比表面积有关:黏土矿物比表面积越大,所提供的氢键位点越多,其对有机质的吸附越强[23].然而,水溶性淀粉在高岭土和蒙脱土上的吸附结果却与此规律不符,原因可能是:(1)高岭土虽然比表面积较小,但以外表面为主,表面利用率较高;蒙脱土尽管有很大的比表面积,但以内表面为主,由于层间距有限(大约为1~2 nm[15]),大分子水溶性淀粉可能难以进入其层间.(2)两种矿物的湿式比表面测定和红外测定结果都表明蒙脱土表面有更多的水分子存在,憎水性的降低可能导致了蒙脱土吸附能力的下降.
2.3 高锰酸钾氧化降解水溶性淀粉
根据预实验可知,水溶性淀粉溶液中加入高锰酸钾氧化后,中间产物会对剩余高锰酸钾在545 nm处的测定产生影响,因此,在氧化实验条件下对剩余高锰酸钾定量时应扣除产物二氧化锰等在545 nm处的影响[24].根据高锰酸钾在300~600 nm范围内的全扫描图(图3(a)),其在419 nm处无紫外光吸收.水溶性淀粉在300~600 nm范围内无紫外光吸收,加入高锰酸钾后的还原产物二氧化锰在419 nm处有紫外光吸收.因此,向过量水溶性淀粉溶液中加入不同量的高锰酸钾,使高锰酸钾完全反应,通过建立545 nm处与419 nm处吸光度的关系(图3(b)),用于氧化实验中基于419 nm处吸光度增加量对545 nm处吸光度的扣除,从而建立修正后545 nm处的吸光度.以往方法或是直接测定545 nm处吸光度,无法得到氧化过程中整个体系内确切的高锰酸钾剩余量[25].或用硫代硫酸钠淬灭高锰酸钾的氧化反应进行间接测定,无法在氧化过程中直接取样测得测定高锰酸钾剩余量[25].上述方法一方面可以得到氧化过程中整个体系内确切的高锰酸钾剩余量,另一方面可以在反应进行过程中直接取样测定,不需要再加入硫代硫酸钠等终止剂,与以往方法相比[25],在方法学上具有一定的创新性.

图3 (a)过量水溶性淀粉溶液与不同量的高锰酸钾反应后紫外光谱图示意;(b)545 nm处吸光度与419 nm处吸光度关系.随着高锰酸钾加入量的增加,419 nm处吸光度不断增大,将419 nm处吸光度与545 nm处吸光度建立关系,发现两者具有很好的正相关.Fig.3(a)The UV spectrum of the reaction of excessive water-soluble starch solution with different amount of potassium permanganate;(b)The relationship between the absorbance at 545 nm and 419 nm.With the increase of the amount of potassium permanganate,the absorbance at 419 nm increased.The relationship between the absorbance at 419nm and the absorbance at 545 nm was established.
利用上述比例关系扣除中间产物的影响后,得到剩余高锰酸钾浓度随时间的变化曲线如图4所示.显然,180 min内,高岭土-水溶性淀粉体系对高锰酸钾的消耗量(1.74×10-2mmol/L)低于蒙脱土-水溶性淀粉体系(1.92×10-2mmol/L),且均低于无矿物体系(2.24×10-2mmol/L).有矿物体系较无矿物体系在相同时间内消耗高锰酸钾的量更少,说明矿物可以通过吸附作用,对水溶性淀粉起到保护作用,降低高锰酸钾对其的氧化作用.未被吸附的水溶性淀粉中的极性基团数量较多,易于被氧化[26];矿物体系中水溶性淀粉的极性基团与矿物表面的活性位点直接作用,使矿物-有机质表面呈现出疏水性,从而减弱了水溶性淀粉与高锰酸钾的反应[26].此外,与蒙脱土相比,高岭土对水溶性淀粉的吸附保护作用更强,可能归因于高岭土对水溶性淀粉较高的吸附量(该条件下,每克高岭土吸附量为2.28 mg,高于蒙脱土的吸附量1.37 mg).

图4 剩余高锰酸钾浓度随时间的变化曲线Fig.4 Change curve of residual potassium permanganate concentration with time
3 结 论
1)高岭土对水溶性淀粉的吸附能力高于蒙脱土,憎水性作用可能是主导吸附机理.
2)矿物可以通过吸附作用保护水溶性淀粉免受高锰酸钾的氧化,且高岭土由于较高的吸附量,具有更强的保护能力.
基于以上结论,我们认为对土壤有机质稳定性的预测和评估,需要更加关注输入量大且易降解的土壤非腐殖质活性组分,发掘优先吸附保护此类有机质的土壤矿物,从分子层面理解矿物晶型结构在土壤非腐殖质活性组分吸附保护中的作用机制,为理解土壤有机质的周转更替,调控土壤有机质的输入,增加土壤肥力、固碳能力和污染物锁定能力提供理论基础.
猜你喜欢
云南化工(2021年5期)2021-12-21
现代塑料加工应用(2021年5期)2021-02-28
云南化工(2020年11期)2021-01-14
山东冶金(2019年1期)2019-03-30
中国环境科学(2018年10期)2018-10-29
腐植酸(2017年4期)2017-11-15
湖南农业科学(2017年10期)2017-03-02
材料科学与工程学报(2016年2期)2017-01-15
天津城建大学学报(2015年5期)2015-12-09
中国塑料(2015年11期)2015-10-14
