
二氧化硅-十八烷基三氯硅烷界面的和频光谱相位测量及其测量精度分析
2021-03-09 10:11刘晓杰李玉琼冯冉冉
光谱学与光谱分析 2021年3期
刘晓杰,徐 帅,李玉琼,靳 刚,冯冉冉,3*
1. 中国科学院力学研究所国家微重力实验室,北京 100190 2. 中国科学院大学工程科学学院,北京 100049 3. 北京大学化学与分子工程学院整合谱学中心,北京 100871
引 言
有机薄膜的界面结构决定了其功能,但关于界面特异性结构的研究还比较有限[1]。和频振动光谱(sum frequency generation,SFG)具有界面选择性和单分子层灵敏性,是研究有机薄膜界面结构的有效方法。SFG能够识别界面分子组成,研究界面分子取向及界面动力学[2-4],在纳米材料、生物传感、微机电等许多研究领域具有广泛的应用前景[5-7]。
和频振动光谱是通过测量样品的二阶非线性极化率(χ(2))来获得界面分子信息的技术,χ(2)是复数,普通SFG仅得到二阶非线性极化率的模,不能直接测量相位信息,需要通过光谱拟合等方法解析χ(2)的相位[8]。此外,如果研究体系复杂,普通SFG强度光谱中共振峰之间以及共振与非共振信号之间存在干涉,导致光谱拟合和分析困难。因此,实验直接测量χ(2)的相位对于解析SFG光谱和界面结构具有重要的意义。
Shen等提出了利用窄带皮秒激光器进行相位SFG测量,通过将样品与已知相位的参考样品干涉来推导二阶非线性极化率的相位[9],但是该方法是单光谱检测,耗时时间长。之后,Tahara[10]和Benderskii[11]等分别提出并实现了宽带SFG的相位测量,弥补了窄带系统光谱采集时间长和信噪比较差的不足。随后,国内外多个研究组发展了多种SFG相位测量方法,包括共线/非共线构型,标准内差/标准外差等测量方法,并将之应用于气/液、气/固界面,体现了相位测量在研究界面光谱和界面结构方面的优势[12-19]。
尽管相位测量SFG光谱具有比普通强度测量SFG光谱的优势,但在实验重复性以及实验设计和界面分析方面仍有一些关键问题没有解决。例如,Shen等首先进行了纯水界面的SFG光谱的相位测量,结果显示低于3 200 cm-1区域的光谱虚部为正,而较高频率3 200~3 600 cm-1的虚部为负[20]。Tahara,Chen,Tian等再次测量了纯水界面的SFG相位,但各研究组之间测得的光谱重复性不高,尤其是关于氢键OH低频区域(<3 200 cm-1)虚部相位是否为正存在争议[17,21-26]。Tahara认为相位标准样品石英导致待测样品的相位误差,改用D2O作为相位标准后,发现纯水界面OH低频区域为零[21]。Allen研究组在不同的时间测量得到的纯水界面的虚部谱也存在约20°的相位误差[26]。最近有报道从实验和理论两个方面系统研究了石英、D2O等标准样品的相位,发现能产生较大体相偶极矩贡献的z-切石英的信号作为参考标准,相位测量准确度高[25]。相位误差会引起光谱较大的变化并误导界面结构分析,因此分析并准确控制误差是相位测量SFG的关键技术。标准样品的选择和样品位置的重复性是SFG相位误差分析的两个重要方面,关于相位标准的选择,相关工作[19,25]已经做了详细的理论和实验分析,而关于样品位置重复性的误差分析,目前还未见有详细的报道。
本工作使用z-切石英作为相位标准,测量了修饰在熔融石英基底上的十八烷基三氯硅烷(octadecyltrichlorosilane,OTS)在C—H振动波段的和频振动光谱,对OTS的相位光谱进行了解析,并讨论了所测样品和参考样品位置的非一致性对相位测量精度的影响。
1 相位测量系统及原理
图1为相位测量系统光路示意图,主要由飞秒钛宝石激光器、光学参量放大器(OPA,自行搭建)、差频器(DFG)、脉冲整形系统、干涉样品台和信号采集系统等组成。钛宝石激光器(Allstrella Coherent,1 kHz,31 fs)产生中心波长810 nm的激光经分束镜功率被分为两部分。其中约2.5 W用于泵浦光参量放大器(OPA)产生信号和闲散光,再经差频系统(DFG)产生可调谐的中红外光。SFG光谱的分辨率取决于可见光的带宽,采用脉冲整形的方法(衍射光栅、透镜、可调狭缝和高反镜) 将钛宝石激光器产生的31fs的激光压缩到带宽约5 cm-1,功率约为12 mW。压缩后的可见光(vis)经延时控制和偏振控制,与中红外光(IR)聚焦到金膜表面(200 nm厚,第一样品台),在满足相位匹配条件下产生和频光(SF1,Local Oscillator,LO)。Vis,IR和SF1经过凹面镜(曲率半径R=25 cm)聚焦到待测样品表面(第二样品台)产生第二束和频光(SF2,Sample)。SF1经过1 mm厚的熔融石英片,与SF2之间产生约1.7 ps的相位延迟,SF1和SF2产生干涉,经光谱仪分光后,用电荷耦合器件(CCD)检测SF1和SF2的干涉光谱。
实验中,先将待测样品(修饰了OTS的熔融石英表面)放置在第二样品台,测量样品和金膜(LO)的SFG干涉光谱,再将z-切石英(参考样品)放置在第二样品台,测量z-切石英和金膜的SFG干涉光谱,经反傅里叶和傅里叶变换等处理过程,得到待测样品表面二阶极化率的实部和虚部光谱。在测量石英信号时,要明确石英晶体的相位定义,选择正确的方向,避免待测样品的相位信息翻转180°[19]。为了保证待测样品和参考样品在测量过程中处于相同的位置和扭转角度,使用了位移传感器定位(Keyence CL-3000,精度: 0.2 μm)。实验中,可见光和中红外光的入射角分别为45°和55°。和频光、可见光和红外光的偏振分别是S,S和P(SSP)。中红外光中心波长约3 450 nm,因此和频光谱采集的中心波长约为650 nm。实验过程中室温控制在20°±0.5°,湿度RH=40%。

图1 相位测量系统光路图
待测样品与本地振荡器(LO)之间的干涉光谱可以表示为
(1)
(2)

2 OTS相位光谱
主要存在两种常见的实验构型,一是可见光和红外光先入射到本地振荡器,再反射到样品表面; 二是可见光和红外光先到达样品表面,经样品反射后再聚焦到本地振荡器。综合考虑表面反射率以及样品和本地振荡器的信号强度等因素,选择了第一种实验构型,实验测量的金膜与OTS、金膜与石英的和频信号强度比分别为20∶1和6∶1,干涉条纹对比度较高,有利于光谱分析。
图2(a)是测量的熔融石英表面OTS与金膜之间的干涉光谱,经反傅里叶变换到时域[图3(a)],再通过滤波函数选取t=1.7 ps处的信号并将滤波后的信号经傅里叶变换回频域,得到复数谱,如图4(a)所示。
类似地,用z-切石英作为参考样品,得到z-切石英和金膜的干涉光谱[图2(b)],与上述数据处理流程类似,相继得到图3(b)和图4(b)。将修饰了OTS的石英和z-切石英的复数谱相比即得到OTS的实部和虚部光谱,如图5所示,黑色和蓝色分别是虚部谱和实部谱,实线是拟合曲线。

图3 (a) 金膜和熔融石英上OTS干涉的时域谱; (b) 金膜和z-切石英干涉的时域谱
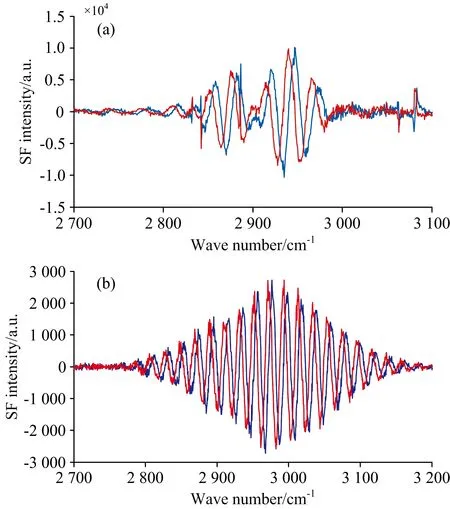
图4 (a)金膜和熔融石英上OTS滤波后的光谱实部(橙色)和虚部(绿色)图; (b)金膜和z-切石英滤波后的光谱实部(红色)和虚部(蓝色)图
在SSP偏振组合下,二阶非线性极化率χ(2)由式(3)给出[19]
(3)

(4)


图5 熔融石英上OTS的实部(蓝色)和虚部(黑色)光谱,实线是拟合曲线
图5所示的OTS虚部光谱中,2 878和2 936 cm-1处的两个正峰,分别指认为末端CH3的对称振动(CH3ss)和费米共振(CH3FR),2 960 cm-1处的负峰指认为CH3的反对称伸缩振动(CH3as),这三个峰的光谱特征和指认与文献相同[19,28]。Wang用内差相位测量方法,将位于~2 891 cm-1并与CH3ss反相位的峰归属为CH2反对称(CH2as)[19]。Aimin等也将在~2 886 cm-1处的峰归属为CH2as。在2 910 cm-1附近也观察到一个负峰,与上述文献比较,约有20 cm-1的偏移,对照文献,可以暂时归属为CH2as。在2 850 cm-1附近还观察到一个负峰,归属为CH2对称伸缩(CH2ss)[29-30],Wang和Amini没有测量到2 850 cm-1处的峰,原因可能是样品制备条件不同影响了OTS的分子排列结构,Wang将石英晶体浸入0.1%OTS溶液(溶剂为200 mL的十六烷,30 mL的四氯化碳和20 mL的混合溶剂)中30 min后在鼓风干燥炉中(60 ℃)烘烤2 h[19]。Ge A的样品制备条件为将石英基底浸入0.15 Wt%的OTS溶液(溶剂为十六烷/四氯化碳/氯仿=80∶12∶8(体积比))中3 h后在110 ℃下烘烤1 h[28]。

(5)
(6)

图6 CH3的与取向角θ之间的关系蓝色: CH3ss; 红色: CH3asFig.6 The relationship between the of CH3and the orientation angle θblue: CH3ss; red: CH3as

图7 相位测量(黑色)和强度测量(红色) 对比光谱图
通过图5中的实部谱和虚部谱可以得到强度光谱|χ(2)|2。与实验直接测得的强度光谱进行对比,如图7所示,通过相位测量和强度测量两种方法得到的强度光谱吻合性较好,验证了相位测量实验结果的可靠性。与强度测量相比,相位测量得到的虚部谱在2 960,2 850和2 910 cm-1处均观察到了明显的峰。更重要的是,相位测量可以直接获得界面分子的取向信息: CH3的三种振动模式的c轴与表面法线的夹角均小于90°,表明CH3的H更多为向上取向且排列有序。
3 位置重复性对测量精度的影响
实验过程中,待测样品与参考样品的相对测量位置会影响相位测量的精度。在替换样品过程中,要求待测样品与参考样品的位置完全一致,否则会引入相位误差。因此,研究了待测样品与参考样品的位置非一致性对相位测量精度的影响。实验中采用了位移传感器和红外指示灯同时监测样品的位置,以保证参考样品和待测样品处于相同的位置。样品放置在一维手动平移台上,通过螺旋调节器控制样品的位置。图8(a)是OTS样品台位位于不同位置时的OTS虚部谱(螺旋调节器示数分别为12.1,12.3和12.73 mm),发现三次实验的虚部谱产生了相对位移,甚至翻转,这是由于样品台前后位置的移动在OTS和石英之间引入了相对相位。
模拟了在待测样品和参考样品之间引入相位误差后虚部谱的变化[图8(b)]。黑色的谱线是实验测量数据,在此基础上分别引入10°,20°,30°,…等相位误差。如图8所示,相位误差的引入会引起振动峰位置的改变、正负号等,导致对光谱的错误解析,例如20°相位的偏移会导致零点位置移动约6 cm-1。严格控制两次样品测量位置的一致性对相位测量非常重要。结合图8中实验和模拟结果,估计了样品台前后位置改变与相位测量角度的关系: 0.2 mm的位移对应于约80°的相位误差,0.43 mm的位移对应于约180°的相位误差,因此2.5 μm的位移对应于1°的相位误差。实验中使用的样品台底座的螺旋调节器的最小测量精度为10 μm,这将引入约4°的相位测量误差。综上,待测样品与参考样品测量位置的相对位移会造成过零点的光谱位置的变化、各振动峰之间的振幅比以及相位谱整体形状的改变等,故需借助位移传感器和指示灯等保证待测样品和参考样品测量位置的一致性。

图8 (a) OTS在三个不同测量位置的相位测量虚部谱: (黑色)12.3 mm,(红色)12.1 mm,(蓝色)12.75 mm; (b)模拟在待测样品和参考样品之间引入10°~90°的相对相位后虚部谱的变化
4 结 论
通过宽带相位敏感和频振动光谱方法对修饰在熔融石英表面的OTS进行了相位测量,从干涉光谱中提取出OTS的相位信息并解析了各振动峰的归属,表明OTS中末端CH3的H更多为向上取向,且在2 850 cm-1处测量到亚甲基的对称伸缩,其虚部为负,2 910和2 960 cm-1处分别测量到亚甲基和甲基的反对称振动峰,表明相位测量相较于强度测量可以获得更丰富的表面信息。同时,通过实验和模拟,研究、分析了待测样品与参考样品的位置非一致性对相位测量精度的影响。结果表明2.5 μm的测量位移对应于1°的相位误差,20°相位的偏移会导致零点位置移动约6 cm-1,引起振动峰位置和符号等的改变,导致对光谱的错误解析,因此待测样品与参考样品位置的一致性是保证相位测量精度的必要条件。本实验研究结果为提高和频振动光谱相位测量的精度与准确性提供了指导,为界面分子表面态的检测与分析、及微小信号的探测提供了有效手段。
猜你喜欢
北京航空航天大学学报(2022年8期)2022-08-31
中学生数理化·高一版(2022年3期)2022-04-15
矿产保护与利用(2022年5期)2022-03-28
中等数学(2021年6期)2021-08-14
数学学习与研究(2020年23期)2020-01-11
电子制作(2018年14期)2018-08-21
卷宗(2016年8期)2016-11-15
中国继续医学教育(2015年1期)2016-01-06
铁道科学与工程学报(2015年5期)2015-12-24
中国光学(2015年5期)2015-12-09
