
基于gp41近膜端外部区域中和表位天然构象的人类免疫缺陷病毒1型疫苗的设计
2021-03-04 10:54:34曹妙苏珊叶玲陆路杨庆来姜世勃
微生物与感染 2021年3期
曹妙,苏珊,叶玲,陆路,杨庆来,姜世勃
1. 复旦大学上海医学院基础医学院教育部/卫健委/医科院医学分子病毒学重点实验室, 上海 200032; 2. 埃默里大学医学院微生物与免疫学系埃默里疫苗中心, 美国亚特兰大 GA30322
人类免疫缺陷病毒(human immunodeficiency virus, HIV)分为 1型和2型,是一种双股正链RNA病毒,隶属于反转录病毒科慢病毒属,可在受体(CD4)和辅助受体(CCR5或CXCR4)的介导下通过膜融合的方式感染免疫细胞,并破坏人体免疫系统,最终引起获得性免疫缺陷综合征(acquired immune deficiency syndrome,AIDS,即艾滋病),甚至导致死亡[1-2]。据联合国艾滋病规划署(United Nations AIDS Program, UNAIDS)报道(1)数据来源于http://www.unaids.org/。,截至2019年底,HIV累计造成约 3 300 万人死亡;全球现存HIV感染者约 3 800 万,每年新感染人数约为170万,每年与HIV感染有关的死亡人数约为69万。
疫苗是控制传染病传播的重要措施。据数学模型统计分析预测,在保持当前诊断和治疗条件不变的情况下,如果从2020年开始70%的人群接种保护率达到50%的HIV疫苗,那么到2035年将保护 1 700 万人免受HIV感染[3]。然而,在过去的40年里,尽管全球在研究HIV疫苗方面做出了大量的努力,但至今仍未能研发出一种有效的HIV疫苗[4]。HIV疫苗研发失败与其包膜蛋白序列高度可变性和高度糖基化的特性有关,即高度可变序列导致病毒对机体产生的HIV中和抗体发生免疫逃逸,而糖基化则为一些保守的表位提供庇护,使其不能被免疫系统识别而难以诱导中和抗体产生[5]。
与HIV-2相比,HIV-1具有更强的毒力和感染性,HIV-1的包膜蛋白gp120和gp41是暴露在病毒颗粒表面的主要抗原,上面有许多能够被广谱中和抗体识别的位点,如gp120的CD4结合位点[6-7]、gp120的V1V2环[8]、gp120的V3聚糖[9]以及位于gp41胞外域C末端的近膜端外部区域(membrane-proximal external region, MPER)[10-12],它们是研发疫苗的关键免疫原。与包膜蛋白上的其他位点相比,gp41的MPER(660LLELDKWASLWNWFD-ITNWLWYIK683)在疫苗设计上具有更大的吸引力。然而,由于MPER免疫原性比较差,旨在诱导MPER特异性中和抗体产生的疫苗策略迄今为止均未成功[13-21]。在HIV-1包膜蛋白的天然构象中,gp41总是被空间位阻比较大的gp120覆盖,导致gp41上的免疫表位,尤其是MPER表位难以被机体的免疫系统识别[22]。去除gp120虽然可以有效暴露gp41上的免疫表位,但单独的gp41或MPER多肽却无法模拟包膜蛋白的天然构象,且诱导产生的抗体亦没有中和活性[21];而在没有gp120存在的情况下,gp41的N末端七肽重复域(N-terminal heptad repeat, NHR)和C末端七肽重复域(C-terminal heptad repeat, CHR)往往会通过强烈的相互作用形成融合后的6螺旋(6-helix bundle, 6-HB)构象[23]。与MPER以及gp41融合前构象相比,6-HB是一种非常强的免疫显性抗原,容易诱导机体产生大量针对它的非中和抗体,从而削弱MPER的免疫原性。因此,如何在充分暴露MPER中和表位的情况下尽可能模拟gp120/gp41的天然构象并避免gp41的免疫显性抗原位点暴露,是亟须解决的问题。
Ye等[24]研究发现,使用空间位阻更小的流感病毒A/Aichi/2/1968(H3N2)血凝素(hemagglutinin,HA)头部结构域HA1替代HIV-1的gp120,与gp41构建成HA/gp41嵌合DNA疫苗,不但可以有效展示位于gp41上的MPER表位,还能正常表达于细胞表面,这在一定程度上模拟了gp120/gp41在病毒包膜上的状态。但该嵌合疫苗也存在一些问题:①使用了同一株流感病毒A/Aichi/2/1968(H3N2)的HA1亚基,在经过反复多次的免疫后,诱导机体产生了大量针对流感病毒HA的抗体,这在一定程度上影响了HIV-1 gp41的免疫原性;②未能解决gp41的NHR和CHR相互作用形成6-HB构象的问题。
本文基于HA/gp41嵌合疫苗的研究,通过在gp41的NHR和CHR引入一系列突变来阻止两者相互作用形成6-HB结构,以维持gp41融合前的天然构象;并以柔性连接子GSGSGSGS替代NHR和CHR之间的环区来尽可能规避gp41上的显性非中和表位,得到gp41的突变体gp41-1605,并用此构建新型HA/gp41嵌合疫苗。为了避免使用相同HA1构建的新型HA/gp41嵌合疫苗诱导机体产生更多针对HA1的抗体,首先使用一系列序列差异较大的流感病毒HA1亚基替代HIV-1的gp120亚基来构建HA/gp41嵌合DNA,并通过体外实验证明这些嵌合DNA能在细胞表面正常表达并被MPER中和抗体结合,同时失去与gp41非中和抗体结合的能力。随后,在新西兰大耳兔上通过肌肉免疫的方式对疫苗的有效性进行评估。本研究期望为HIV-1疫苗的研究提供有益的数据参考。
1 材料与方法
1.1 实验材料
1.1.1 多肽所有多肽(纯度>98%)均由KarebayBiochem公司(中国宁波)合成。
1.1.2 细胞和病毒株HeLa细胞和U87细胞来源于美国模式培养物集存库保藏中心(American Type Culture Collection,ATCC)。MT-2细胞、HIV-1实验室适应株 HIV-1ⅢB(KJ925006.1)及表达T7聚合酶的牛痘病毒VTF7-3均来源于美国国立卫生研究院艾滋病试剂项目(NIH AIDS Reagent Program)。
1.1.3 蛋白及抗体识别N36和C34多肽的多克隆抗体NY364由美国纽约血液中心杜兰英博士惠赠[25];6-HB特异性小鼠单克隆抗体(NC-1)[26]、特异性识别NHR三聚体的鼠单克隆抗体18D3[27]、识别HIV-1 gp41 loop中显性抗原位点的小鼠单克隆抗体F240[28]、MPER特异性小鼠单克隆非中和抗体(5F3)[29]以及MPER特异性人单克隆中和抗体(4E10和2F5)[10, 29-30]均来自美国国立卫生研究院艾滋病试剂项目;甲型流感H3N2(A/Aichi/2/1968)HA和抗甲型流感H3N2(A/Aichi/2/1968)HA(HA1亚单位)的兔多克隆抗体均购自北京索莱宝生物公司;特异性识别甲型流感H3N2(A/Aichi/2/1968)HA的免疫兔血清Jose由David Steinhauer博士惠赠[31]。
1.1.4 实验动物新西兰大耳兔(9月龄雌兔,3~3.5 kg),在复旦大学公共卫生临床中心饲养。
1.2 实验方法
1.2.1 酶联免疫吸附实验(enzyme linked immunosorbent assay, ELISA)检测NHR和CHR突变多肽相互间形成6-HB的情况以N36和C34分别作为NHR和CHR的代表多肽,于37 ℃孵育30 min形成6-HB,将其作为阳性对照。将可以结合N36和C34多肽的NY364抗体包被于96孔聚苯乙烯板中,4 ℃过夜;将浓度为5 μmol/L的被检多肽(N36突变多肽或C34突变多肽)与5 μmol/L 的检测多肽(C34突变多肽或N36突变多肽)以1∶1的体积比混合,37 ℃孵育30 min;在包被NY364抗体的96孔板中加入封闭液[含体积分数为1%的脱脂牛奶的0.05%吐温-20磷酸盐缓冲液(phosphate buffered saline-Tween-20,PBS-T)],置于37 ℃封闭2 h后,每孔加入多肽混合物 50 μL,再置于 37 ℃孵育1 h;洗去多肽,每孔加入对HIV-1 gp41的6-HB结构具有特异性的单克隆抗体NC-1(浓度5 μg/mL)50 μL,37 ℃孵育1 h;洗去NC-1抗体,每孔加入辣根过氧化物酶(horseradish peroxidase, HRP)标记的兔抗鼠单克隆抗体50 μL(用PBS-T 1∶4 000 稀释),37 ℃孵育1 h;洗去HRP标记的单克隆抗体后每孔加入 50 μL ELISA底物显色液;待显色明显后,加入 50 μL 浓硫酸终止液终止反应,用酶标仪测定各孔在 450 nm 处的吸光度值(A450)。
1.2.2 ELISA检测N36突变多肽形成三聚体的能力以原始多肽N36作为阳性对照。将浓度为 5 μmol/L 的N36突变多肽包被于96孔聚苯乙烯板中,置于4 ℃过夜;用封闭液在37 ℃封闭2 h后,每孔加入HIV-1的NHR三聚体特异性单克隆抗体18D3(浓度为 5 μg/mL)50 μL[27],37 ℃孵育 1 h;洗去18D3抗体,每孔加入50 μL用PBS-T以 1∶4 000 稀释的 HRP标记的兔抗鼠单克隆抗体,37 ℃孵育1 h;洗去HRP标记的单克隆抗体后每孔加入 50 μL ELISA底物显色液;待显色明显后,加入 50 μL 浓硫酸终止液终止反应,用酶标仪测定各孔在450 nm处的吸光度值(A450)。
1.2.3 构建包含不同gp41突变的新型HA/gp41嵌合DNA将用于体外表达验证的HA/gp41嵌合DNA构建到pBlueScript Ⅱ KS载体中[32];动物免疫用的HA/gp41嵌合DNA用pCAGGS表达[33]。具体方法如下:以流感病毒A/Aichi/2/1968(H3N2)的HA质粒为模板,分别以GCCACCAT-GAAGACCATCATTGCT和ATTGCGCCGAA-TAGGCCTCGAGTTTGTTTCTCTG作为上、下游引物,在3′末端或HA1编码序列引入XhoI限制性酶切位点(划线部分为酶切位点);以HIV-189.6的包膜蛋白质粒为模板,分别以CAAACTCGAG-GCATCGGCGCCGTTAATGGC和TCAAGGG-CAGCAGGTCTGGAA作为上、下游引物,在gp41的5′末端引入XhoI限制性酶切位点(划线部分为酶切位点),通过聚合酶链反应(polymerase chain reaction,PCR)分别对HA1基因和gp41基因进行扩增。将HA1基因的扩增产物通过酶切、连接克隆到pBlueScript Ⅱ KS载体中[32]。根据碧云天定点突变试剂盒设计引物,在gp41的NHR区和CHR区引入相应的突变;通过酶切、连接将突变后的gp41克隆到带有HA1的pBlueScript Ⅱ KS载体中,在gp41上构建包含不同突变位点的HA/gp41嵌合DNA:HA/gp41-1686、HA/gp41-1605、HA/gp41-GSL、HA/gp41-EKL、HA/gp41-CM、HA/gp41-NM(见表1)。

表1 HA/gp41疫苗的构建
1.2.4 ELISA检测包含不同gp41突变的HA/gp41嵌合DNA用ELISA对HA/gp41嵌合DNA在细胞表面的表达及对HIV-1中和/非中和抗体的识别分别进行检测,检测方法如下[32]:转染前将 1×104个HeLa细胞铺在96孔平底细胞培养板中,隔夜生长至密度为90%左右;用重组痘苗病毒VTF7-3感染细胞,在感染复数(multiplicity of infection,MOI)值为10的情况下感染2 h后,用构建于pBlueScript Ⅱ KS载体的疫苗质粒转染细胞,以pBlueScript Ⅱ KS空载体作为阴性对照。转染后18 h,用PBS将96孔板中的细胞洗1遍,以2%甲醛固定后用牛血清白蛋白(bovine serum albumin, BSA)阻断进行封闭。洗去BSA后,将细胞与特异性识别流感病毒株A/Aichi/2/1968(H3N2)HA1的兔免疫血清(Jose)[31]常温孵育1 h;洗去血清后,加入HRP标记的山羊抗兔IgG抗体,常温孵育45 min;洗去HRP标记的单克隆抗体后每孔加入50 μL ELISA底物显色液;待显色明显后,加入50 μL浓硫酸终止液终止反应,用酶标仪测定各孔在450 nm处的吸光度值(A450)。
在检测嵌合HA/gp41疫苗抗原对MPER特异性中和抗体、非中和抗体、6-HB特异性抗体以及免疫显性表位非中和抗体的识别时,一级抗体分别用MPER特异性中和抗体(4E10和2F5)[10, 29-30]、6-HB特异性抗体(NC-1)[26]、MPER非中和抗体(5F3)以及gp41 环区免疫显性表位特异性非中和抗体(F240)[28]进行检测。
1.2.5 构建包含不同HA1亚单位的HA/gp41-1605嵌合DNA对gp41上包含不同突变位点的HA/gp41的特性进行检测后,在HA/gp41-1605的基础上,使用不同流感病毒[A/Shanxi/10/2006(H5N1)、A/California/07/2009(H1N1)、A/Qinghai/1/2005(H5N1)和A/Puerto Rico/8/1934(H1N1)]的HA1亚基取代HA/gp41-1605的HA1结构域,构建包含不同HA1的嵌合DNA:HA/gp41-1605、HA(SX)/gp41-1605、HA(CA)/gp41-1605、HA(QH)/gp41-1605以及HA(PR8)/gp41-1605;构建用于动物免疫的包含不同HA的HA/gp41-1605 DNA时,将pBlueScript Ⅱ KS载体替换为pCAGGS载体。
1.2.6 ELISA检测包含不同HA1亚单位的HA/gp41-1605嵌合抗原用ELISA对HA/gp41-1605嵌合抗原在细胞表面表达及对HIV-1中和/非中和抗体识别分别进行检测,检测方法同1.2.4小节。一级抗体分别用特异性识别流感病毒株A/Aichi/2/1968(H3N2)HA1的免疫血清Jose、中和抗体(4E10和2F5)及非中和抗体(F240和5F3)进行检测。
1.2.7 动物免疫所有DNA疫苗均采用磷酸铝作为佐剂,多肽疫苗均采用Sigma佐剂系统(Sigma Adjuvant System, SAS)。每次免疫前均通过耳缘静脉采集血样,免疫时所有兔子均通过后腿外侧肌肉接种疫苗,每只兔子每次接受的DNA剂量为500 μg,多肽剂量为50 μg。免疫前一天将抗原与佐剂混合制成免疫用疫苗,4 ℃保存。具体方法如下。①DNA疫苗:用0.9%生理盐水将嵌合DNA稀释至0.5 mg/mL,再按1∶1的体积比加入磷酸铝佐剂(Sigma公司),持续吹打混匀5 min以上,置于 4 ℃ 保存备用。②多肽疫苗:用二甲亚砜(Dimethyl sulfoxide, DMSO)溶解10E8-4P多肽,再用0.9%生理盐水将多肽稀释至0.25 mg/mL,使用注射器将多肽加到2 mL 45 ℃预热的SAS佐剂中,旋涡混匀至少5 min,暂存于4 ℃备用。
1.2.8 免疫兔血清中特异性抗体检测使用ELISA检测免疫兔血清中的HA、gp41 CHR、gp41 NHR、6-HB、MPER以及10E8表位特异性抗体的产生情况,具体如下:将多肽(或流感病毒HA蛋白)稀释至5 μg/mL,用碳酸盐缓冲液包被至96孔聚乙烯板中,4 ℃过夜;用封闭液封闭后,每孔加入50 μL用PBS-T稀释100倍的免疫兔血清,置于37 ℃孵育1 h后,加入HRP标记的羊抗兔IgG抗体,常温孵育45 min;每孔加入50 μL ELISA底物显色液;待显色明显后,加入50 μL浓硫酸终止液终止反应,用酶标仪测定各孔在450 nm处的吸光度值(A450)。检测血清中的HA抗体时,以特异性识别流感病毒株A/Aichi/2/1968(H3N2)HA1的抗体作为阳性对照;检测血清中gp41 CHR和NHR特异性抗体时,以NY364抗体作为阳性对照。
在检测6-HB特异性抗体时,首先将5 μg/mL 6-HB特异性抗体NC-1包被于96孔聚乙烯板中,4 ℃ 保存过夜;并将原始N36和C34多肽按1∶1比例混合,置于37 ℃孵育30 min;待用封闭液对 NC-1 抗体进行封闭后,加入N36和C34多肽混合物,37 ℃孵育1 h后,每孔加入50 μL用PBS-T稀释100倍的免疫兔血清,以NY364抗体作为阳性对照,37 ℃孵育1 h,加入HRP结合的羊抗兔IgG抗体,常温孵育45 min后每孔加入50 μL ELISA底物显色液;待显色明显后,加入50 μL浓硫酸终止液终止反应,用酶标仪测定各孔在450 nm处的吸光度值(A450)。
1.2.9 免疫兔血清对HIV-1ⅢB的中和活性检测免疫兔血清用无血清的改良Eagle培养基(Dulbecco’s modified Eagle medium,DMEM)梯度稀释,在96孔平底细胞培养板中每孔加入50 μL稀释血清,并加入50 μL含100 TCID50的HIV-1ⅢB病毒株,于37 ℃、体积分数为5% CO2的细胞培养箱中孵育30 min,设置病毒对照孔和细胞对照孔。30 min后,每孔加入100 μL 1×104MT-2细胞,过夜感染,然后用含10% 胎牛血清(fetal bovine serum, FBS)的新鲜DMEM更换培养上清液。感染后第4天,待观察到病毒对照孔出现明显病变后,从每孔中收集50 μL培养上清液,与等体积的5% Triton X-100混合,并通过ELISA检测p24抗原。
2 结果
2.1 NHR区的N36-M3、N36-M4、N36-M6和N36-M9突变有助于维持gp41融合前的天然构象
为了鉴定NHR上能够阻止gp41形成融合后的6-HB结构并维持gp41融合前非6-HB天然构象的关键氨基酸,本研究基于之前关于NHR和CHR上位点突变的报道,在N36的CHR结合位点引入1~2个氨基酸突变[23, 34-38],并通过带电荷的谷氨酸(E)和赖氨酸(K)替换N36上亮氨酸(L)和异亮氨酸(I)等疏水性氨基酸,在N36多肽内部引入E-K盐桥以稳定其α-螺旋构象并阻止N36和CHR相互作用,得到10条N36突变多肽(N36-M1~N36-M10,见表2)。为了筛选NHR上能够阻断6-HB结构形成的有效突变位点,使用ELISA检测了10条N36突变多肽形成三聚体构象和与原始C34多肽作用形成6-HB构象的能力。结果显示,N36上这些突变都能有效阻断N36与原始C34形成6-HB构象(见图1A)。并且在这10条N36突变肽中,N36-M3(L568P/V570E)、N36-M4(L568P/W571E/Q575K)、N36-M6(L556K/V570P)和N36-M9(T569P/Q575A)还能形成N36三聚体构象(见图1B)。

A: The interaction between NHR mutant peptides and CHR peptide C34 to form 6-HB conformation. B: The ability of N36 mutant peptide to form NHR-trimer.

表2 NHR和CHR突变多肽序列
2.2 CHR区中C34-M1、C34-M3和C34-M4突变有助于维持gp41融合前的天然构象
在C34突变多肽的设计上,主要通过在C34与NHR结合的位点引入E-K盐桥(如W631K/I635E、Y638K/I642E和L645E/S649K)来阻断C34与NHR的相互作用,以达到阻止6-HB形成的目的。最终,得到了5条C34突变多肽(C34-M1~C34-M5,见表1),并使用6-HB特异性抗体(NC-1),通过ELISA检测这些C34突变多肽能否与原始N36多肽相互作用形成6-HB结构。结果显示,C34-M1(W631K/I635E)、C34-M3(Y638K/I642E)和C34-M4(L645E/S649K)均不能与N36形成6-HB结构(见图2)。

The interaction between CHR mutant peptides and natural NHR peptide N36 to form 6-HB conformation detected by ELISA.
2.3 NHR区N36-M6突变和CHR区C34-M1、C34-M3突变组合能够维持gp41融合前的天然构象
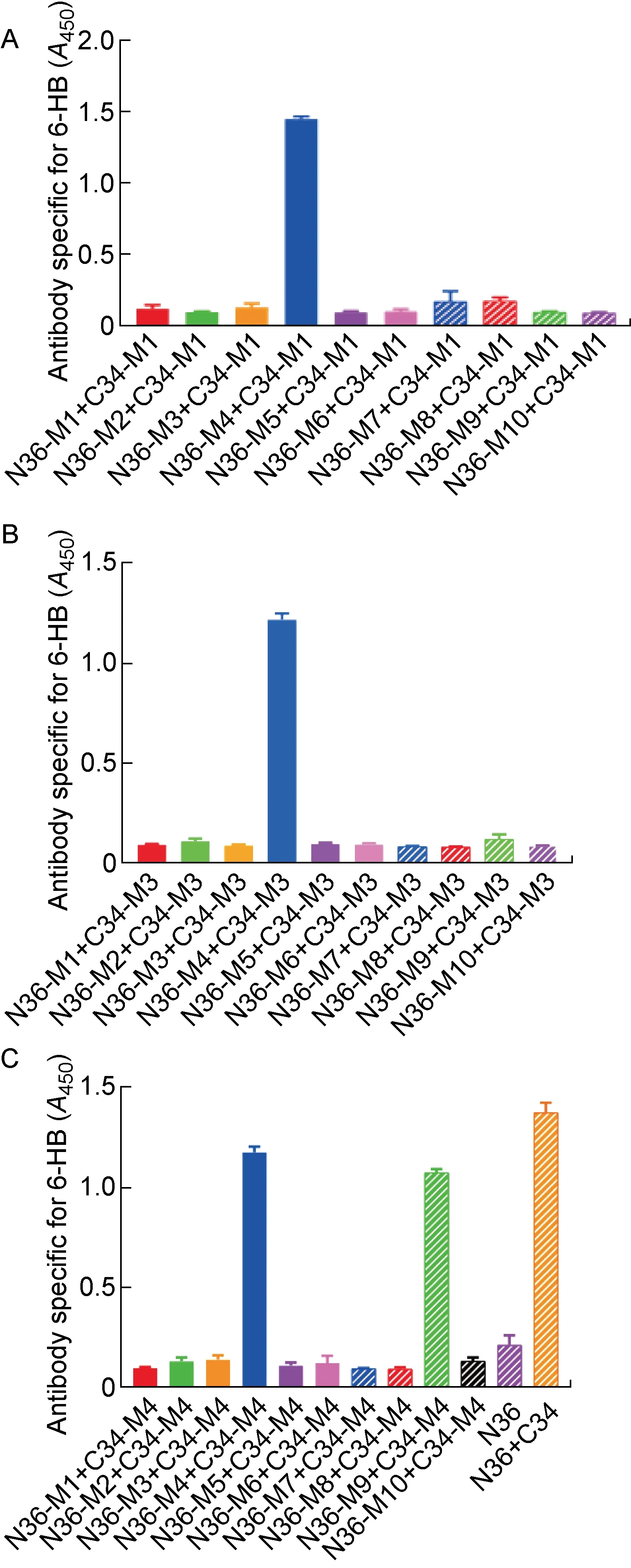
上述结果表明,在C34突变多肽中,只有C34-M1、C34-M3和C34-M4这3条突变多肽在与原始N36多肽作用后未形成6-HB结构,因此进一步检测这3条C34突变多肽能否与10条N36突变多肽形成6-HB结构。结果显示,C34-M1和C34-M3只能与N36-M4形成6-HB构象,而C34-M4可以与2条N36突变多肽(N36-M4、N36-M9)形成6-HB构象(见图3),表明以C34-M1和C34-M3为代表的W631K/I635E和Y638K/I642E突变可以作为嵌合疫苗构建过程中CHR区突变的候选者。考虑到在N36突变多肽中, 仅N36-M3、N36-M4、N36-M6以及N36-M9能够维持三聚体构象, 而N36-M4和N36-M9还有可能与C34突变多肽形成6-HB,因此,排除N36-M4和N36-M9两种突变策略。由于原始N36多肽在PBS中的溶解性不佳,进一步检测10条N36突变多肽在PBS中的溶解性后,发现所有N36突变多肽在PBS中的溶解性都比原始N36多肽更好,而其中N36-M2(L556K/L568P)和N36-M6(L556K/V570P)具有最高的溶解度(见表3)。考虑到N36-M2无法模拟天然的三聚体结构,最终确定将以N36-M6(L556K/V570P)为代表的突变多肽应用到构建HA/gp41嵌合疫苗的策略中。
C34-M1 (A), C34-M3 (B) and C34-M4 (C) forming 6-HB conformation with N36 mutations detected by ELISA.

表3 NHR及其突变多肽在PBS中的溶解性
2.4 HIV-1 gp41上不同位点的突变对HA/gp41 DNA疫苗抗原性的影响
为了确定用于动物免疫的嵌合DNA的具体构建方案,须明确HIV-1 gp41上不同位点的突变对HA/gp41 DNA疫苗抗原性的影响差异。首先,以流感病毒株A/Aichi/2/1968(H3N2)HA1亚基取代HIV-1包膜蛋白gp120,并在gp41上引入T569P突变以增加疫苗的表面稳定性,构建了6株在gp41上包含不同突变的HA/gp41嵌合DNA,克隆到pBlueScript Ⅱ KS质粒载体中。6株嵌合DNA构建策略如下:①HA/gp41-1686在gp41区域仅包含T569P突变;②HA/gp41-1605在gp41区域不仅包含T569P突变,而且包括NHR(L556K/V570P)和CHR(W631K/I635E/Y638K/I642E)的突变,并且用免疫性弱且更灵活的GSGSGSGS接头序列替代了含有免疫优势表位的gp41环;③HA/gp41-GSL除包含T569P突变外,还使用GSGSGSGS取代环;④HA/gp41-EKL除包含T569P突变外,还使用更坚固的EAAAKEAAAKEAAAK接头取代环;⑤HA/gp41-CM仅含有CHR突变(W631K/I635E/Y638K/I642E)及T569P突变;⑥HA/gp41-NM仅含有NHR(L556K/V570P)突变及T569P突变(见表1)。
随后,用ELISA检测细胞表面抗原,以确定HA/gp41-1686、HA/gp41-1605、HA/gp41-GSL、HA/gp41-EKL、HA/gp41-CM和HA/gp41-NM嵌合蛋白是否能正确折叠并成功转运到细胞表面。如图4A所示,所有HA/gp41嵌合疫苗抗原都能在细胞表面被流感病毒毒株A/Aichi/2/1968(H3N2)HA1的特异性兔免疫血清(Jose)识别,说明这些疫苗抗原均能在细胞内合成后被运输到细胞表面;进一步使用ELISA检测细胞表面嵌合蛋白对MPER特异性中和抗体(4E10)和6-HB特异性非中和抗体(NC-1)的反应。结果表明,HA/gp41-1605保留了对4E10的反应性(见图4B),而无法被NC-1识别(见图4C),说明HA/gp41-1605能够更好地展示MPER表位,且不会形成6-HB构象。此外,与其他HA/gp41嵌合蛋白相比,HA/gp41-1605也无法被非中和抗体(5F3和F240)识别(见图4D、4E),说明HA/gp41-1605可能是一个比较合适的候选嵌合疫苗。
2.5 包含不同甲型流感病毒(influenza A virus,IAV)HA1亚基的HA/gp41-1605嵌合抗原对相关抗体的反应
为了避免具有相同流感病毒HA1亚基的嵌合疫苗反复免疫诱导机体产生更多针对HA1的抗体,依据HA/gp41-1605的构建方式,使用不同流感病毒HA1[A/Shanxi/10/2006(H5N1)、A/California/07/2009(H1N1)、A/Qinghai/1/2005(H5N1)、A/Puerto Rico/8/1934(H1N1)]构建HA(SX)/gp41-1605、HA(CA)/gp41-1605、HA(QH)/gp41-1605以及HA(PR8)/gp41-1605嵌合DNA(见图5A),并以相同的方式检测这些嵌合抗原在细胞表面的表达以及被HIV-1中和抗体和非中和抗体识别的情况。结果显示,替换了HA1之后,嵌合蛋白都不能被流感病毒毒株A/Aichi/2/1968(H3N2)HA1的兔免疫血清(Jose)识别(见图5B);同时,新构建的嵌合蛋白都能被MPER特异性中和抗体(4E10和2F5)识别(见图5C、5D)。以上说明新构建的嵌合蛋白能在细胞体系上正常表达,且保留了展示MPER表位的能力。此外,与HA/gp41-1605类似,替换了HA1的嵌合蛋白也不能被gp41特异性非中和抗体5F3以及F240识别(见图5E、5F)。

Construction of HA/gp41-1605 DNA with different HA1 subunits (A); Recognition of the cell-expressed HA/gp41-1605 chimeric DNA vaccine antigen by HA[A/Aichi/2/1968(H3N2)]-specific antibody (Jose, B); MPER-specific neutralizing antibody (4E10, C); MPER-specific neutralizing antibody (2F5, D); MPER-specific non-neutralizing antibody (5F3, E); gp41 loop-specific non-neutralizing antibody (F240, F). HA/gp41-1686 is the positive control antigen for the antibodies tested, while pBlueScript II KS vector is the negative control (see the “Control” in each figure).
2.6 序贯免疫包含不同IAV HA1的HA/gp41-1605嵌合DNA疫苗可有效避免机体产生针对HA的抗体
为了增强免疫效果,使用HA/gp41-1605嵌合DNA疫苗进行序贯免疫,同时在DNA免疫前后分别使用之前开发的包含4个串联10E8表位的10E8-4P多肽进行加强免疫。将9只新西兰大耳兔随机分为3组(每组3只),具体免疫程序如表4所示。第1组为实验组(1~3号),前5次分别使用含有不同HA1的HA/gp41-1605嵌合DNA疫苗免疫,最后采用10E8-4P多肽进行加强免疫;第2组为对照组(4~6号),每次均采用HA/gp41-1686(包含同一HA1且gp41未经阻断6-HB形成突变改造的嵌合DNA疫苗)进行免疫,最后2次仍然采用10E8-4P多肽进行加强免疫;第3组(7~9号)与第1组采用相同的免疫原,只是在免疫程序上首先使用多肽免疫,再使用DNA疫苗免疫。

表4 动物免疫程序
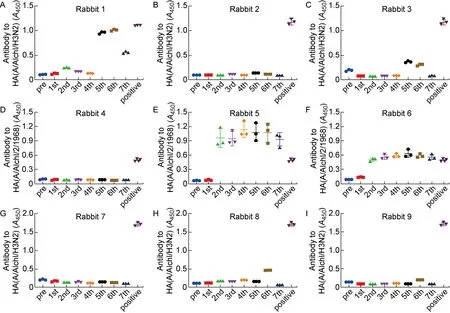
在经过7次免疫后,首先检测免疫兔血清对HA1[A/Aichi/2/1968(H3N2)]抗原的识别能力。如图6所示,第2组中的5号、6号兔在第2次免疫后就产生了针对HA1(H3)的抗体,说明一直免疫同种HA1的HA/gp41嵌合蛋白确实存在诱导产生特异性HA1抗体的问题。而在第1组和第3组使用包含不同HA1嵌合DNA疫苗进行序贯免疫的6只大耳兔中,仅有1只在第5次免疫后产生了针对HA1(H3)的抗体,说明使用不同HA1作为嵌合疫苗头部的免疫策略对于规避更为显性的HA1免疫原性确有一定作用。
Anti-HA antibody in immunized rabbit sera with 1: 100 dilution was detected by ELISA. HA antibody against A/Aichi/2/1968(H3N2) HA was used as positive control.
2.7 序贯免疫包含不同IAV HA1亚基的 HA/gp41-1605嵌合DNA疫苗能够有效避免产生针对gp41上非中和表位的抗体
进一步检测免疫兔血清与gp41 CHR、gp41 NHR和6-HB的结合情况。9只大耳兔均未产生针对HIV-1 gp41 NHR结构域以及6-HB结构的抗体(见图7B、7C),说明疫苗策略确实能够有效避免gp41上非中和位点产生抗体。此外,在对照组(第2组)的1只大耳兔血清中检测到了针对HIV-1 gp41 CHR结构域的抗体(见图7A),而第1组与第3组的6只大耳兔均未产生针对gp41上其他表位的抗体,在一定程度上证明了新构建的嵌合疫苗能更好地避免其他非中和表位产生抗体。

Recognition of HIV-1 gp41 CHR domain (A), NHR domain (B) and 6-HB conformation (C) by antibodies in rabbit antisera, respectively. NY364 antibody was used as positive control.
2.8 包含不同 HA1 的 HA/gp41-1605 嵌合DNA疫苗的序贯免疫可诱导MPER特异性抗体产生,但对HIV-1不具有中和作用
检测免疫兔血清与MPER多肽以及10E8表位多肽的结合情况,发现除实验组(第1组)1号兔在第7次免疫后产生了针对MPER多肽的抗体外,其他兔血清中均未检测到MPER特异性抗体(见图8A)。所有兔血清中均未检测到针对10E8表位的抗体(见图8B),也未检测到对HIV-1ⅢB感染具有抑制作用的中和抗体(见图9)。

Recognition of HIV-1 gp41 MPER peptide (A) and 10E8 epitope (B) by antibodies in rabbit antisera.

MT-2 cells were infected by 100 TCID50 HIV-1 in the presence or absence of rabbit antisera at different dilutions. The p24 antigen was detected by sandwich ELISA.
3 讨论
针对艾滋病疫苗设计,位于HIV-1 gp41的MPER因具有以下特性而表现出极大的优势。①MPER序列在不同亚型的HIV-1毒株中相对保守,其诱导产生的中和抗体不易使HIV-1发生免疫逃逸。相比之下,gp120在表面存在许多具有免疫优势的可变区,可能诱导机体产生大量没有中和活性的抗体[39-40];此外,gp120是一个高度糖基化蛋白,这些复杂而密集的糖基化位点可能对gp120上保守的抗原表位形成遮盖,导致其难以被机体的免疫系统识别[41]。②MPER在HIV-1感染以及病毒和细胞的膜融合过程中发挥着重要作用[42]。有研究发现,删除HIV-1的MPER上17个氨基酸(665WASLWNWFDITNWLWYI683)后,HIV-1在细胞间传播以及进入靶细胞的能力将受到影响[43];而MPER上659LLELDKWASLW671序列的删除也影响了HIV-1感染靶细胞后合胞体的形成[44]。③多个从HIV-1感染者体内分离出的单克隆抗体被证明具有中和HIV-1的能力,如2F5、4E10和10E8就能分别通过识别MPER的656NEQELLELDK-WASLWN671、671NWFDITNWLWYIK683以及664DKWASLWNWFDITNWLWYIK683序列,进而中和57%~67%、~98% 和~98%的HIV-1临床株[12]。
迄今为止,针对MPER开发的HIV-1疫苗未能诱生高效、广谱的HIV-1中和抗体,这可能是由MPER的免疫原性差以及其中和位点未能充分暴露,且在gp41的环区和6-HB结构中存在的免疫优势位点可诱导产生大量非中和抗体,进一步削弱MPER中和位点免疫原性引起的[45]。先前的研究发现,使用IAV的HA1亚基替代HIV-1的gp120与gp41构成的HA/gp41嵌合疫苗抗原,不仅能在一定程度上模拟gp120/gp41包膜蛋白的结构,还能有效暴露gp41上的免疫表位。但HA1和gp41的环及融合后6-HB结构的强免疫原性可能削弱了MPER的免疫原性,导致未能诱导产生高效的MPER特异性中和抗体[24]。本研究通过在gp41的NHR和CHR引入突变来稳定其融合前非6-HB结构的天然构象,用免疫原性弱的氨基酸片段替换环,并使用空间位阻较小的、不同流感病毒毒株的HA1亚基替换gp120,以期更好地暴露gp41中MPER抗原表位。结果显示,表达于细胞表面的HA/gp41-1605嵌合蛋白能够被MPER特异性中和抗体4E10和2F5所识别,表明其能有效展示MPER的中和抗体结合表位。此外,有研究报道,NHR两段α-螺旋之间的L568-V570区域存在一个转角结构,并且有研究发现在N36的L568位点引入脯氨酸(P)突变能够降低6-HB结构的稳定性[37],并显著增强MPER区4E10表位的免疫原性[38]。在设计gp41突变体时,发现在L568-V570的转角结构中引入一个P突变的NHR突变多肽不能与原始C34多肽形成6-HB结构,表明引入P突变可能对阻断6-HB结构的形成格外重要。
之前,对于10E8-4P的构象分析发现,10E8-4P表现出排列良好的串联螺旋构象,能够使10E8表位中的关键氨基酸残基有效暴露。与仅包含一个10E8表位的多肽10E8-1P相比,10E8-4P不仅显示出更好的抗原性,而且诱发了针对HIV-1假病毒的中和抗体应答[46]。本研究在进行疫苗免疫原性评估时,采用了两种不同的策略。第一,先用嵌合HA/gp41-1605疫苗对新西兰大耳兔进行序贯免疫以激活所有针对HIV-1 gp41的B细胞;再用包含10E8表位的10E8-4P多肽进一步进行加强免疫,以期刺激产生MPER特异性抗体。第二,先用10E8-4P多肽进行免疫以激活针对天然或非天然构象的MPER抗体,再用包含MPER表位天然构象的嵌合HA/gp41-1605疫苗进行序贯免疫,以期得到针对天然构象的中和抗体。
结果表明,两种免疫策略在诱导中和抗体产生方面并无明显差异。嵌合疫苗HA/gp41-1605的序贯免疫的确能有效避免机体产生针对HA的抗体和针对gp41上免疫优势区的非中和表位抗体,但是却不能诱导产生具有中和活性的MPER特异性抗体。可能的原因如下。①在自然状态下, MPER具有天然的柔性,能够以不同的构象与不同的中和抗体结合[12, 47-48],虽然融合前的非6-HB构象可能是gp41诱导中和抗体产生的天然构象,但在gp41的NHR和CHR引入突变后可能也影响了MPER的构象。而最近的研究也发现,MPER不是稳定的三聚体结构,而是适应于融合后结构变化的动态片段[49],这在一定程度上增加了模拟MPER天然构象进行疫苗研发的难度。②有研究发现4E10、10E8和2F5 等MPER特异性中和抗体既能结合病毒包膜上的MPER,又能结合合成的MPER多肽。但是,如果突变去除互补决定区的H3(complementarity-determining region-H3, CDR-H3)后,这些抗体只能识别MPER多肽,而不再识别膜上的MPER,并且同时失去了中和活性[50-52]。目前,发现的MPER抗体都有很长的CDR-H3,可能需要长时间的体细胞高频突变才能实现,而本研究的免疫时间可能还不够长,故诱导的抗体只能识别MPER多肽,不能识别膜上的MPER,因此没有中和活性。③针对MPER的广谱中和抗体大多具有自身反应性,可能削弱抗体的中和作用[53-54]。例如:疫苗刺激可能产生MPER特异性广谱中和抗体的pre-B细胞,在骨髓中发育时,其中大多数可能通过克隆缺失和受体编辑而被去除,因此不能发育为未成熟的IgM + B细胞;仅有少数脂质(或其他自身抗原)反应性B细胞能够成功从骨髓迁移到次级淋巴器官并分化为可产生抗体的B细胞。只有少量无反应性细胞可以移动至次淋巴器官,因此产生MPER特异性广谱中和抗体十分困难。
此外,有研究认为磷脂可能也在MPER特异性抗体对HIV-1中和方面发挥作用。比如:MPER的大部分氨基酸都包埋在病毒膜中[55-56],而MPER上与4E10抗体结合的3个至关重要的疏水性氨基酸(W672、F673和L679)也都被包埋在脂质膜中,仅关键氨基酸T676暴露于病毒膜磷脂头部[55]。大量通过脂质体模拟脂膜的MPER疫苗研究发现,脂膜确实可以通过促进天然构象的形成来调节MPER的结构,并改善其免疫原性[57-58]。本研究在体外实验中证明嵌合DNA确实能够表达于细胞表面,这在一定程度上模拟了MPER在病毒脂膜上的状态。不过,表达于细胞表面的HA/gp41-1605是否与HIV-1的包膜蛋白具有相同的构象并模拟MPER的天然结构尚不清楚。而且,本研究的DNA疫苗是通过局部注射的方式进行免疫,可能无法激活大规模的免疫反应,从而导致无法诱导产生具有中和活性的HIV-1抗体。
本研究发现,表达于细胞表面的HA/gp41-1605嵌合蛋白能够被MPER中和抗体4E10和2F5识别,但是在使用HA/gp41-1605 DNA疫苗免疫家兔后,获得的血清中的MPER抗体却不能识别包含4E10和2F5表位的10E8-4P多肽,说明即使将gp41稳定在融合前的天然构象,MPER仍可能处于动态变化之中。因此,在今后的疫苗设计中,可能须考虑如何进一步稳定MPER构象。冠状病毒疫苗的相关研究可能会带来一些启示,例如目前进入Ⅲ期临床的新型冠状病毒mRNA-1273疫苗通过在冠状病毒刺突(spike,S)蛋白上引入2个脯氨酸突变(S-2P)来稳定S蛋白融合前的构象,以增强其免疫原性[59-61]。因此,在HA/gp41-1605的基础上,可能也可以采用该策略在gp41的MPER引入突变来稳定MPER构象,进一步增强疫苗的免疫原性。
此外,裸露的DNA疫苗也面临转染效率不高和易被机体降解的风险[62-63],这可能是DNA疫苗诱导中和抗体的免疫原性较差的一个重要原因。当前,越来越多的研究将HIV-1疫苗平台聚焦于病毒样颗粒(virus-like particles, VLPs)或重组病毒疫苗,它们或能通过模拟天然状态下MPER与脂质的结合特征以提高疫苗的免疫原性,或能够提高DNA疫苗的转染效率和诱导中和抗体的能力。而许多以VLPs或重组病毒为平台的MPER疫苗,也诱导产生了具有一定中和活性的抗体[24, 64-65]。
综上所述,在基于MPER的疫苗研发方面,除了中和抗体表位序列外,MPER的天然构象及其在天然状态下所处的脂膜环境,甚至是疫苗形式可能也须予以考虑[66]。
猜你喜欢
中学化学(2024年2期)2024-06-17 04:01:47
遵义医科大学学报(2023年4期)2023-05-05 05:05:16
中国兽药杂志(2021年7期)2021-08-13 02:37:22
艺术品鉴(2020年5期)2020-07-27 02:43:08
丹青少年(2017年2期)2017-02-26 09:10:51
中国免疫学杂志(2017年1期)2017-01-17 04:53:25
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
畜牧兽医学报(2015年3期)2015-07-05 08:22:53
医学研究杂志(2015年6期)2015-07-01 17:41:11
应用化工(2014年7期)2014-08-09 09:20:23
