
神经元核内包涵体病诊治分析:附一例伴眼睑痉挛的病例报道
2021-03-03 09:31:10王晶晶崔丽丽王焕苗江永张祥建
实用心脑肺血管病杂志 2021年2期
王晶晶,崔丽丽,王焕,苗江永,张祥建
神经元核内包涵体病(neuronal intranuclear inclusion disease,NIID)是一种慢性进展的神经系统退行性疾病,其典型特征是患者中枢、周围神经系统神经元和胶质细胞及内脏细胞内存在嗜酸性透明包涵体,并因此而得名[1],亦可称为神经元核内透明包涵体病(neuronal intranuclear hyaline inclusion disease,NIHID)或核内包涵体病(intranuclear inclusion body disease,INIBD)。NIID的临床表现复杂多变,主要包括:锥体系和锥体外系症状、小脑共济失调、痴呆、肌无力、感觉障碍以及自主神经功能受损(如呕吐、膀胱功能障碍、晕厥和瞳孔缩小)等;影像学特征是皮髓质交界区弥散加权成像(diffusion-weighted imaging,DWI)高信号,其对NIID的诊断具有重要参考价值;目前广泛应用的确诊方式是皮肤活检[2]。随着NIID相关基因的发现,基因检测也成为其重要诊断手段之一[3]。发病率低及临床表现多样使得NIID临床诊断困难,目前仅百余例相关报道,其中亚洲以日本报道居多,以成年人发病多见,国内报道较少见。本文报道1例以痴呆、眼睑痉挛为主要表现的成人NIID患者,并通过分析其临床表现及进行文献复习,以期加强临床医生对NIID的认识。
本文创新点/不足:
神经元核内包涵体病(NIID)是一种慢性进展的神经系统退行性疾病,属少见病,国内报道较少。其临床表现复杂多变,主要包括锥体系和锥体外系症状、小脑共济失调、痴呆、周围神经损伤及自主神经功能障碍等,但尚未见关于出现眼睑痉挛的报道,本文丰富了该疾病的临床特点。NIID以皮髓质交界区绸带样弥散加权成像(DWI)高信号为特征影像学表现,本文报道的患者颅脑磁共振成像(MRI)首次提示脑干受累。眼睑痉挛的病理机制尚不清楚,现有证据不能排除其他病因所致,这使得结论存在一定的局限性,仍需要进一步研究。
1 病例简介
患者,男,70岁,高中文化水平,主因“认知功能下降10年,运动迟缓3年余,加重1个月”于2019-12-09就诊于河北医科大学第二医院。患者10年前出现记忆力减退,以近记忆力减退为主,症状进行性加重,并于3年前因记忆力减退就诊于北京某医院,颅脑磁共振成像(magnetic resonance imaging,MRI)示:脑白质、双侧丘脑、脑桥及双侧桥臂多发斑点、斑片状长T1、长T2异常信号及液体衰减反转恢复(fluid attenuated inversion recovery,FLAIR)高信号,部分皮质下U纤维显示欠清;DWI示部分外缘稍高信号,见图1A;未予特殊治疗。患者3年来无明显诱因出现发作性意识模糊、不识家人并无法进行语言沟通3次,伴行走困难、四肢僵直、小便失禁,不伴发热、抽搐及肢体无力,每次发作后给予改善循环、营养神经等治疗后好转,发作间期仍有认知障碍慢性进展,并出现过2次迷路走失。2年前患者突然出现无法自主睁开双眼,分开眼睑时有较大阻力,不伴吞咽困难、面肌及下颌肌紧张,遂就诊于眼科医院,考虑为眼睑痉挛,给予局部注射肉毒毒素治疗,期间共注射4次,每次在1~2 d内完全缓解,疗效可维持6个月。患者9个月前再次发生运动迟缓,走路前倾,止步困难,故于河北医科大学第二医院神经内科就诊。颅脑MRI示:双侧大脑半球皮质下及脑干可见对称性T1等/稍低信号,T2高信号,FLAIR高信号;DWI示皮髓质交界区高信号,见图1B。脑电图示背景活动异常:基本节律7 Hz,θ波,低中幅,对称。神经传导速度测定示:(1)右正中神经、右尺神经末端潜伏期延长,运动及感觉传导速度均减慢;(2)左腓总神经复合肌肉动作电位波幅降低,传导速度减慢;(3)右腓总神经、双胫神经传导速度减慢;(4)右正中神经、右尺神经、双胫神经“F”波潜伏期延长。肌电图示:双胫前肌呈慢性神经源性损伤。给予多巴胺受体激动剂、营养神经、改善循环等治疗后症状明显好转。患者2个月前出现小便失禁。1个月前,患者不明原因出现认知障碍明显加重,表现为不能记起刚刚发生的事情,丢三落四,计算不能,自理能力明显下降,同时伴有运动迟缓、走路前倾及止步困难,未行特殊治疗;半个月前再次发生眼睑痉挛,影响正常生活,遂行肉毒毒素治疗,症状完全缓解。为进一步治疗认知障碍及行走困难就诊于河北医科大学第二医院神经内科。
患者既往有慢性支气管炎病史数年,其父亲有疑似痴呆症状史。个人史、婚姻史无特殊。入院查体:体温36.7 ℃,脉搏70次/min,呼吸频率17次/min,血压132/78 mm Hg(1 mm Hg=0.133 kPa)。意识清楚,少言,记忆力、定向力差,计算力下降。双侧瞳孔等大等圆,直径2 mm,双侧瞳孔直接、间接对光反射迟钝,瞬目减少,轻度上睑下垂。四肢肌力Ⅴ-级,四肢肌张力稍增高。指鼻试验欠稳准,跟膝胫试验欠配合。余未见明显异常。患者住院后完善相关检查,颈部血管超声示:双侧颈动脉内中膜不均匀增厚伴斑块(多发),右侧锁骨下动脉斑块形成。肺部CT示:双肺支气管炎,局部支气管扩张伴感染;双肺散在炎症;双肺上叶前段小结节;双肺下叶散在含气囊腔;心脏增大。颅脑MRI示:皮髓质交界区DWI高信号,白质弥漫性T2及FLAIR高信号,脑干斑片状FLAIR、DWI高信号,双侧小脑中脚FLAIR高信号,胼胝体压部DWI高信号,见图1C、图2。蒙特利尔认知评估量表(Montreal Cognitive Assessment,MoCA)评分6分,简易智力状态检查量表(Mini-mental State Examination,MMSE)评分10分。由于患者家属拒绝行皮肤活检,根据患者临床表现及影像学表现,临床诊断为NIID。入院后给予改善循环、营养神经、抗炎等治疗后,患者锥体外系症状明显好转,但认知功能未见明显改善。于2019-12-17出院后半年内症状无明显波动。
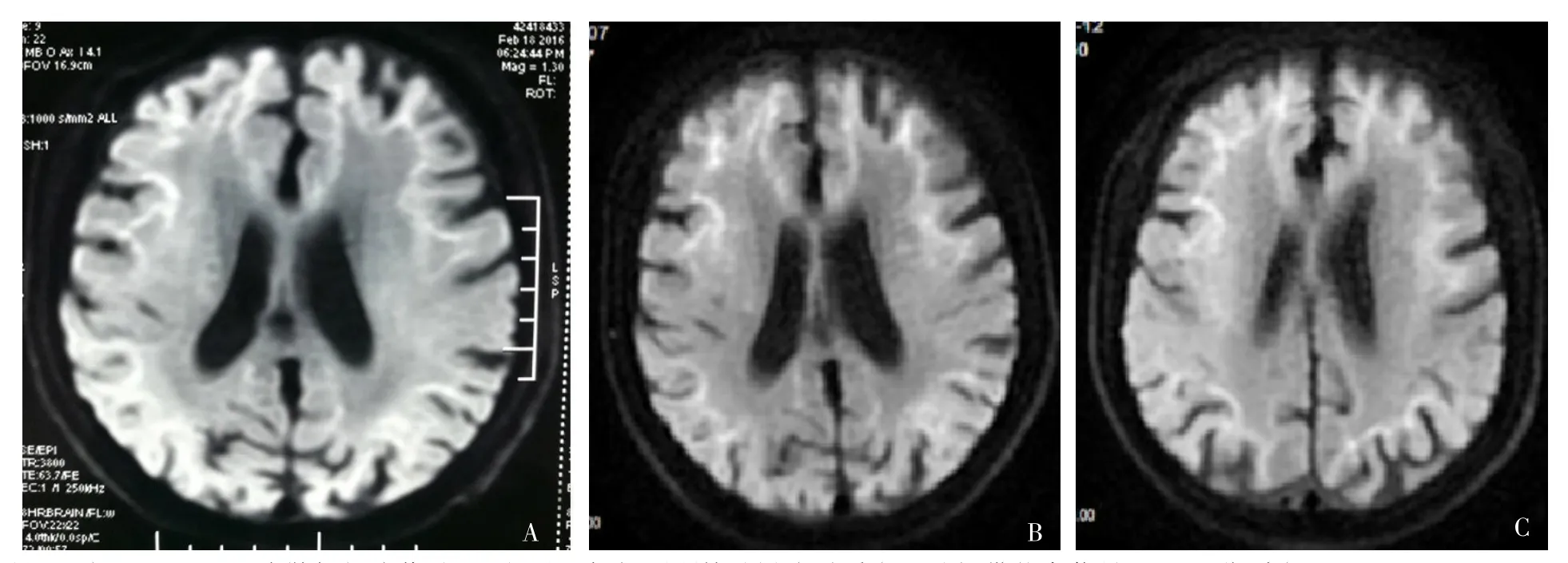
图1 患者颅脑DWI结果Figure 1 Craniocerebral DWI results of patients
2 文献检索
以“核内包涵体、神经元核内包涵体病、神经元核内透明包涵体病”为关键词,检索万方数据知识服务平台、中国知网、PubMed数据库1968年1月—2020年7月公开发表的NIID病例。共检索到国内外文献报道43篇[2-44],结合本例,共纳入34例成人散发型NIID患者及来自11个家庭的29例成人家族型NIID患者,其临床资料详见表1。对其临床表现进行分析:成人散发型NIID患者主要表现为亚临床的周围神经损伤、慢性进展的认知障碍及自主神经功能障碍,除此之外,以震颤、癫痫样发作、脑炎样发作、中风样发作(肌无力或感觉障碍)为主要表现的病例报道也日渐增多。成人家族型NIID患者则以慢性进展的肌无力、自主神经功能障碍(膀胱功能障碍或假性肠梗阻表现等)为主要表现;其中,认知障碍、膀胱功能障碍在两型中均主要表现为慢性进展,而消化道症状、精神行为异常、意识障碍则主要表现为间歇性发作,见表2。肌无力在成人家族型NIID中表现为由下向上慢性进展的对称性肢体无力,在成人散发型NIID中主要表现为无临床症状的周围神经脱髓鞘,少数患者表现为类卒中样的单侧肢体无力。两型中合并眼部症状者均较少,除1例表现为进展性上睑下垂的患者外[44],余多以视力下降或瞳孔缩小为主要表现,本例患者则以眼睑痉挛为特点。
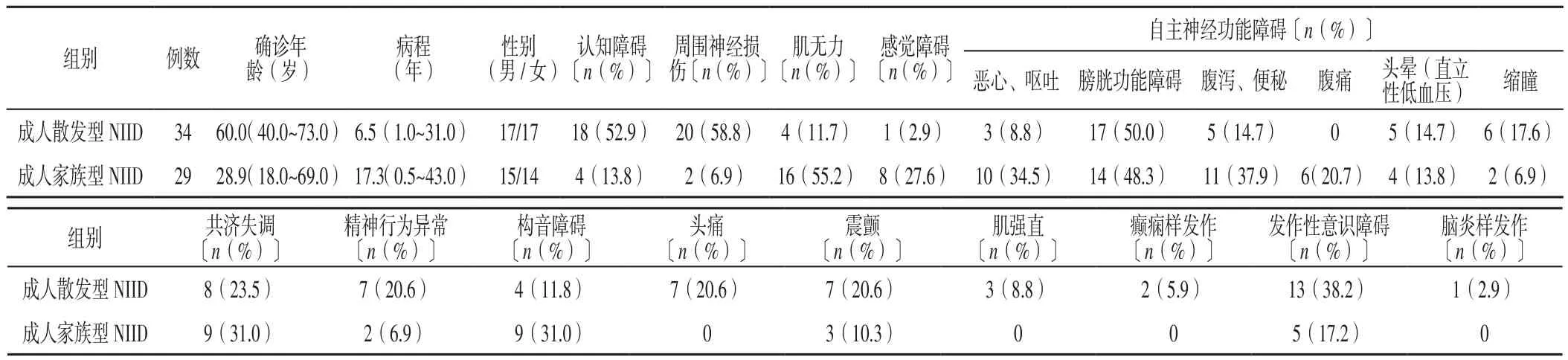
表1 63例成人散发型、家族型NIID患者临床资料Figure 1 Clinical data of 63 sporadic and familial NIID patients
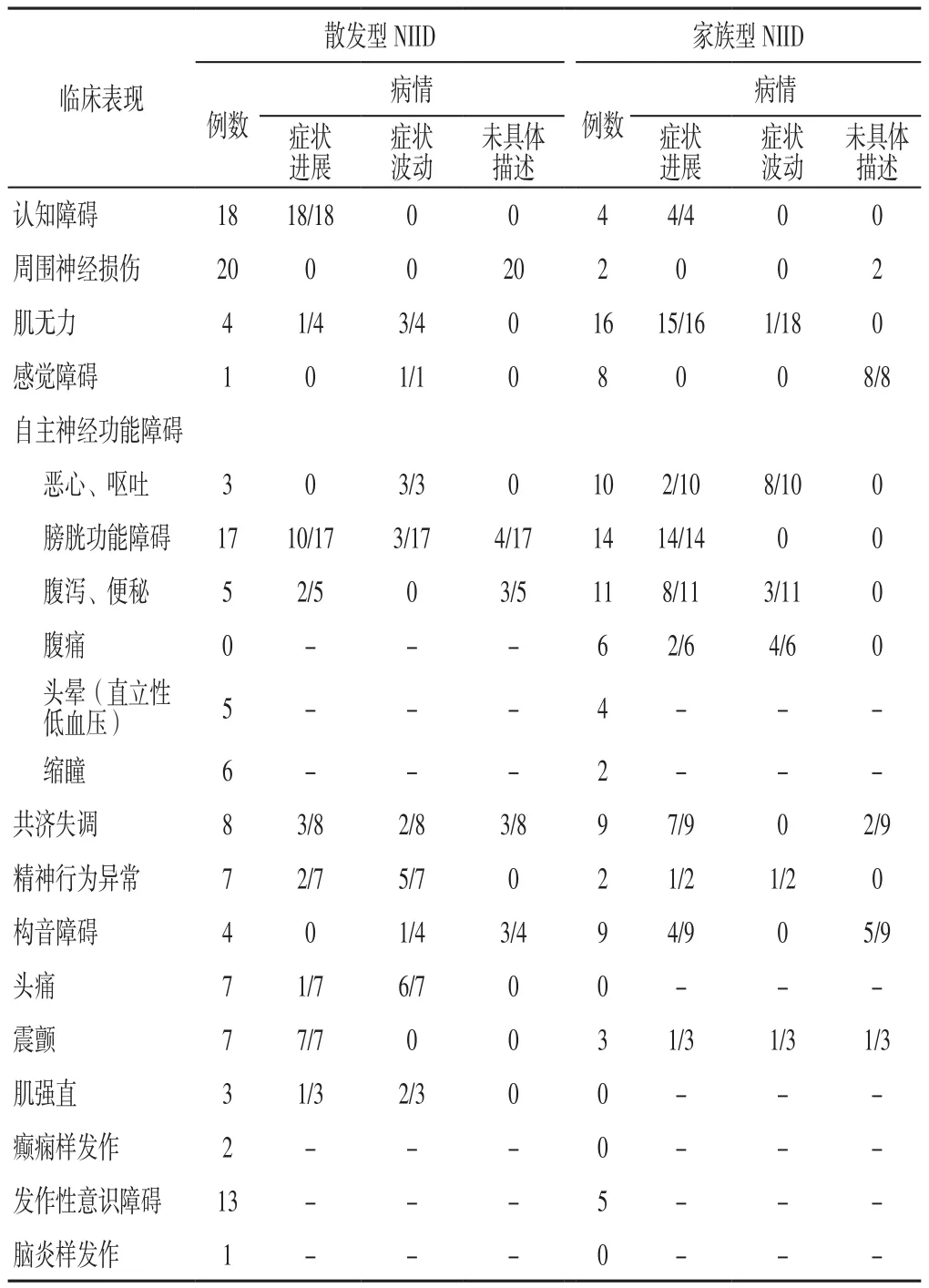
表2 不同临床表现成人散发型、家族型NIID患者病情进展情况(n/N)Table 2 Disease progression of adult sporadic and familial NIID patients with different clinical manifestations
3 讨论
3.1 NIID病理改变及病理生理机制 NIID的主要病理改变为弥漫性神经元丢失及神经元核内包涵体(neuronal intranulear inclusions,NIIs)形成。NIIs是1~10 μm大小的球形透明包裹体,由丝状及颗粒状聚集体组成,无膜结构[45]。免疫组化检查显示,NIIs呈泛素强阳性,且大多含有泛素相关蛋白〔P62、泛素化调控系统的负性调节因子1(negative regulator of ubiquitin-like protein-1,NUB1)、类泛素蛋白修饰分子1和类泛素蛋白修饰分子2〕[46]。泛素-蛋白酶体系统是降解细胞内蛋白的重要途径之一,主要涉及两个连续过程:多个泛素片段与底物耦联,产生多泛素降解信号;下游26S蛋白酶复合物对标记蛋白破坏,并由去泛素化酶(deubiquitinase,DUBs)催化释放出可重复使用的游离泛素[47]。NIIs中泛素和泛素相关蛋白的存在可能与泛素-蛋白酶体系统受损导致一些异常蛋白在细胞核内积聚有关[48]。核体(the nuclear body,NB)是细胞核内的特定核结构域,早幼粒细胞白血病蛋白(PML)作为NB的重要成分之一参与泛素介导的蛋白降解过程[45,49]。NB可被外部感染、激素或肿瘤等应激状态造成的细胞压力激活,从而发挥清除蛋白的作用[50]。NIID中的NB结构异常,呈未活化状态,可能影响了蛋白的清除[49]。NIIs对特异性抗体1C2(识别异常扩增的聚谷氨酰胺)呈阳性反应[51],且NIIs中存在含多聚谷氨酰胺的蛋白(ataxin-1和ataxin-3)[42,52],说明NIID与多聚谷氨酰胺疾病可能存在共同的发病机制[53]。在募集并隔离多种核内外蛋白形成NIIs的过程中,募集蛋白丧失生理功能,进一步导致神经元功能障碍。目前已于NIIs中发现FUS、视神经磷酸酶、ubiquilin 2、含缬氨酸蛋白、SIGMA-1受体和FIG4等多种蛋白,其在膜转运、信号传导及蛋白降解中起重要作用[54-57]。G蛋白耦联受体26(G protein-coupled receptors 26,GPR26)是介导细胞外的刺激向细胞内转化的膜蛋白之一,亦在NIIDs中呈免疫阳性[58]。尽管NIIs的异常出现可能是造成NIID的罪魁祸首,但有研究显示NIIs与神经元丢失呈负相关[59],所以NIIs的形成对神经元可能没有直接毒性作用,而是在泛素-蛋白酶体系统水解过程中被激活,以避免异常蛋白的过度积累。
3.2 NIID临床分型及表现 NIID发病年龄差异较大,婴儿或少年时期发病者约占 2/3[45]。TAKAHASHI-FUJIGASAKI[45]根据发病年龄将NIID分为婴儿型、青少年型及成人型;根据遗传规律将NIID分为散发型和家族型。本研究文献分析表明,散发型NIID发病年龄为40.0~73.0岁,平均发病年龄为60.0岁,平均病程为6.5年,家族型NIID患者发病年龄为18.0~69.0岁,平均发病年龄为28.9岁,平均病程为17.3年。成人散发型NIID主要症状是痴呆、周围神经损伤、膀胱功能障碍、发作性意识障碍和共济失调,成人家族型NIID则突出表现为肌无力、感觉障碍、自主神经功能障碍、共济失调及构音障碍。5岁以前发病者称为婴儿型NIID,共济失调和构音障碍为其首发症状,另有不自主运动、早产及癫痫发作,病程不超过10年;青少年型NIID通常以人格及行为改变为主要特征,伴锥体系及锥体外系症状进行性加重,病程通常为10年以上。
迄今为止,还没有关于NIID患者出现眼睑痉挛的报道。眼睑痉挛被视为局灶性肌张力障碍的一种形式,病因尚不清楚,过去认为主要原因是基底神经核功能障碍[60]。但一项回顾性研究对48例合并局灶性脑部病变的眼睑痉挛患者进行分析发现,多个脑区(丘脑、脑干、基底核、中脑、小脑和皮质)均有受累[61]。由以上研究打破原有观点,眼睑痉挛被认为是一种网络障碍,网络内1个脑区的功能障碍或不同脑区之间的异常通信均有可能导致眼睑痉挛[60]。本例患者同时出现皮质、脑干、小脑及胼胝体病变,涉及以上网络障碍模型中的多个脑区,为发病提供病理基础。但由于其病理机制尚不明确,不能排除其他病因所致,使得结论存在一定的局限性,仍需要进一步研究。
3.3 NIID影像学表现 颅脑MRI对NIID的诊断具有重要参考价值,皮髓交界DWI高信号和脑白质弥漫性T2、FLAIR高信号是NIID的特征性表现[2]。DWI高信号首先出现在额叶皮髓质交界区,并逐渐向后发展到顶叶和枕叶,但不累及深部白质[2]。YOKOI等[62]研究显示,皮髓交界DWI高信号和白质FLAIR高信号分别与病理海绵状改变和弥漫性髓鞘苍白密切相关。海绵状改变仅限于靠近U纤维近端的皮质下病变,可能导致水扩散失活。DWI上的异常信号可一直存在,也可随时间延长而消失[23]。异常信号消失的原因可能与水肿的消退[2]或神经元丢失和胶质增生有关[63]。SUGIYAMA等[64]在多例NIID患者小脑半球蚓旁和小脑中脚发现了异常FLAIR高信号,但在2例脆性X相关性震颤和共济失调综合征(fragile X-associated tremor and ataxia syndrome,FXTAS)的报道中发现了同样的小脑异常信号[65]。另有多例患者在疾病早期仅显示胼胝体压部异常DWI高信号,提示这可能是NIID患者早期影像学特征[66]。本研究文献分析显示,患者均以皮髓质交界区DWI高信号为特征,而本例患者同时具备皮髓质交界区DWI高信号,白质弥漫性T2及FLAIR高信号,脑干斑片状FLAIR、DWI高信号,双侧小脑中脚FLAIR高信号及胼胝体压部DWI高信号。其中,脑干受累少见。
3.4 NIID诊断与鉴别诊断 NIID的确诊手段及标准逐渐演变,自1968年通过尸检确诊首例NIID[1]后,尸检、神经活检及直肠活检作为确诊手段被广泛应用,并由此发现NIIs广泛分布于大脑皮质、基底核、脑干、脊髓及周围神经的神经元、胶质细胞及各种内脏器官的体细胞。在婴儿型及青少年型NIID中,NIIs多见于神经元,成人型NIID更常见于胶质细胞[56]。2011年,SONE等[67]在NIID患者皮肤的脂肪细胞、成纤维细胞和汗腺细胞中发现了嗜酸性泛素阳性和p62阳性的NIIs,使得皮肤活检成为该疾病的确诊手段[19],并在2016年提出了成人型NIID的诊断标准[2]。近期在基因层面对NIID研究较多,于多例患者中均发现了NOTCH2NLC基因CGG三核苷酸重复序列的扩展[68-69],这为诊断NIID增加了基因手段[3]。通过基因测序发现NIID并不罕见,且可能是白质脑病的主要病因之一[70],目前NIID尚无统一诊断标准,但应肯定皮肤活检及基因检测的诊断价值,并重视影像学的辅助诊断价值。本例患者拒绝进行皮肤活检及基因检测,依据临床表现及影像学检查给出临床诊断:成人散发型NIID。
在NIID的鉴别诊断中应考虑多种疾病,包括Shy-Drager综合征(Shy-Drager syndrome,SDS)及FXTAS等。SDS是多发于中老年的原因不明的中枢神经多系统变性疾病,以自主神经功能障碍为主要临床表现,同时也可伴有小脑、脑干或锥体束损伤表现,为多系统萎缩的一类(多系统萎缩-A型),基本病理表现为神经元缺失、胶质细胞增殖,特异性标志是少突胶质细胞包涵体,无特异影像学表现[71]。不同于SDS,NIID患者中枢神经系统、周围神经系统和体细胞(包括皮肤)中均存在嗜酸性包涵体,皮肤活检可鉴别两者。FXTAS是一种进行性神经退行性疾病,该病主要累及脆性智力低下1号(fragile X mental retardation 1,FMR1)基因前突变等位基因(55-200 CGG重复)男性携带者,其特征为运动性震颤、共济失调、帕金森病症状和执行功能障碍[72]。在FXTAS患者的神经元、神经胶质细胞和体细胞中可观察到嗜酸性泛素阳性核内包涵体,目前,仅凭组织病理学很难区分NIID和FXTAS,需进行基因检测。
3.5 NIID治疗 目前尚无特异性治疗方法可治愈或延缓NIID进展,针对复杂临床表现者,治疗以营养神经、改善循环和给予多巴胺类似物、受体激动剂、大剂量激素、丙种球蛋白及手术等对症治疗为主。经上述治疗后,相关症状大多可在短时间内得到控制,但长期治疗的有效性仍待商榷。
综上所述,NIID是一种慢性进展的神经系统退行性疾病,具有特征性的病理和影像学表现及多样的临床表现。成人散发型NIID主要表现为亚临床的周围神经损伤、慢性进展的认知障碍及自主神经功能障碍,成人家族型NIID则以慢性进展的肌无力、自主神经功能障碍(膀胱功能障碍或假性肠梗阻表现等)为主要表现。其中,认知障碍、膀胱功能障碍在两型中均主要表现为慢性进展,而消化道症状、精神行为异常、意识障碍则主要表现为间歇性发作。两型中合并眼部症状者均较少,多以视力下降或瞳孔缩小为主要表现,尚未见合并眼睑痉挛者。全面认识NIID患者临床特征,对疾病的早期诊断具有重要意义。NIID目前尚无特效治疗方法,以对症治疗为主,早期治疗可提高患者生活质量,改善患者预后。
作者贡献:王晶晶进行文章的构思与设计、文献/资料收集与整理,撰写论文;崔丽丽、苗江永进行文章的可行性分析;王焕、苗江永、张祥建进行论文、英文的修订;王晶晶、苗江永负责文章的质量控制及审校,对文章整体负责、监督管理。
本文无利益冲突。
猜你喜欢
阅读(科学探秘)(2021年12期)2021-05-30 10:11:06
反射疗法与康复医学(2017年4期)2017-06-05 09:26:30
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15 12:47:42
中国医疗美容(2015年2期)2015-07-19 10:11:59
中国医疗美容(2015年2期)2015-07-19 10:11:59
中国医疗美容(2015年1期)2015-07-12 10:06:39
中国医疗美容(2015年1期)2015-07-12 10:06:35
中国医学科学院学报(2015年5期)2015-03-01 04:03:46
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
中医研究(2014年10期)2014-03-11 20:29:41
