
高铁酸钾氧化去除弱碱性地下水中低浓度石油类污染物实验研究
2021-03-01 10:01李韵文王文灿李佩佩
四川环境 2021年1期
张 兴,李韵文,王文灿,杨 敏,李 丽,李佩佩
(1.中铁科学研究院有限公司成都分公司,成都 611756;2.西南交通大学,成都 611756)
前 言
高铁酸钾是一种较洁净、具有强氧化能力的氧化剂。当用于氧化去除有机污染物时,其氧化最终产物为CO2和Fe3+,是自然环境中常见无机物质,已被广泛用于水环境污染治理中,氧化去除水中的有机污染物。李洪枚[1]研究了高铁酸钾氧化去除含苯胺黑药废水,结果表明,随反应时间延长和高铁酸钾投加量增大,废水中苯胺黑药去除率逐渐提高;较低的废水起始pH(5.85)和较高反应温度(30℃)都有利于苯胺黑药的去除;在废水起始pH=8.0、30℃、苯胺黑药初始质量浓度40mg/L、高铁酸钾初始质量浓度0.8 g/L条件下反应30 min,苯胺黑药去除率可达98.38%。武秀文等[2]研究用高铁酸钾在pH=4的酸性条件下,氧化污水中的苯酚类污染物,其实验得到初始浓度为100mg/L的苯酚类有机物的氧化去除率达到90.2%,氧化反应时间0.5小时,显示出高铁酸钾的强氧化能力。吉芳英[3]等研究针对高铁酸钾氧化去除以分散油粒态存在的萘、菲和芘等芳烃类有机污染物,其研究显示,在pH=7.1的弱碱性溶液中,对初始浓度110mg/L 3种有机物,氧化去除率可分别达到96%、84%和61%,氧化反应时间5~10min。何世鼎、王凯凯等[4]研究了高铁酸钾在污水处理过程中对有机物的降解特性,研究表明在酸性条件下,高铁酸钾投加量增加更有助于有机物的降解,投加质量浓度增加到50 mg/L后,高铁酸钾对水中类酪氨酸、类富里酸类有机物均有较好的去除效果。
作为一种较洁净的氧化剂,目前高铁酸钾也被用于石油类污染地下水的原位治理。付新建、化勇鹏[5-6]等研究了高铁酸钾投加量、反应时间和pH等因素对地下水有机物降解效果的影响,在一般石油类污染地下水中,随渗流弥散输运的石油类污染物的稳定浓度较低,一般不高于5mg/L。窦文龙等人[7]论述了原位修复技术可渗透反应墙不仅经济有效、节约土地资源,而且对环境干扰小,不占用地面空间,可以实现地下水的持续原位修复,因此,有着良好的应用前景,综述了零价铁PRB去除污染物的作用效果及机理;同时也分析了目前PRB存在的一些问题,对可渗透反应墙未来的发展方向进行了展望。本次研究将高铁酸钾注入构建在污染含水层的PRB渗透反应墙中,直接氧化去除地下水中石油类污染物,不同于前述实验研究的一般高浓度石油类污染物的氧化去除,在地下水石油类污染治理中,研究在地下水溶液弱碱性环境中、高铁酸钾对低浓度石油类污染物的氧化规律、石油类污染物氧化去除率和氧化速率以及氧化反应产物小分子有机酸累积的影响等。
1 实验部分
1.1 实验材料与仪器
实验采用0#轻质柴油模拟石油类污染物,配制石油类污染地下水。其烃分子中碳原子数在10~22个之间,统计分子式为C15.37H27.87O0.67S0.007N0.001,分子量约为214.40g/mol。根据刘泽龙[8]等对0#轻质柴油的烃类组份分析,其组份分类构成见表1。

表1 0#轻质柴油烃类组分构成
水样采用地下水配制,原水取自生活饮用水供水厂的地下水,pH=7.23,总碳酸盐碱度104mg/L,COD<5mg/L,石油类污染物为未检出。高铁酸钾氧化剂采用纯度99.99%高铁酸钾[K2FeO4]固体颗粒。
实验采用200mL烧杯,JJ-6B六联异步恒速电动搅拌器。pH测定采用MP511型pH计。
石油类污染物浓度按照《水质石油类和动植物油类的测定红外分光光度法(HJ 637-2012)》推荐的方法测定。
1.2 试验测试过程
污染水样制备:取200mL水样在烧杯中,称量加入0#轻质柴油,用JJ-6B六联异步恒速电动搅拌器,以200rpm转速连续搅拌不小于1h,再静止约1h。测定石油类污染物浓度,作为实验水样。配制4个浓度实验水样,浓度分别为5.02mg/L、2.05mg/L、1.01mg/L和0.52mg/L。
高铁酸钾氧化剂溶液制备:纯水溶解高铁酸钾[K2FeO4]固体颗粒,配制高铁酸钾氧化剂溶液。在弱碱性溶液中,高铁酸钾离解反应见式(1)。
(1)
高铁酸钾溶解初期有小量[Fe6+]被氧化还原,释放出O2和OH-1,溶液的pH值会上升,至pH>8.0后,Fe6+的还原停止[9-10]。在溶解高铁酸钾时,保持溶液pH>8.0,配制高铁酸钾氧化剂稳定。低浓度石油类有机物氧化速率较低,氧化反应时间长,在氧化反应实验过程中,为避免被氧还原,高铁酸钾保持在稀溶液状态。实验按表1中石油类组分完全氧化所需高铁酸钾量的理论计算值的1∶1、1∶2、1∶3投加高铁酸钾。
实验取实验水样200mL在烧杯中,放置于JJ-6B六联异步恒速电动搅拌器上,以200rpm转速搅拌模拟完全混合状态。待烧杯中水样搅拌混合稳定后,加入高铁酸钾氧化剂溶液连续搅拌。烧杯中石油类污染物被高铁酸钾氧化,其浓度和溶液pH连续下降,每5小时取水样测定石油类污染物浓度和pH值。直至连续3次取样测得的石油类污染物浓度和pH值趋于稳定,不再显著下降时反应结束。
实验以每个浓度水样为一组,石油类污染浓度与高铁酸钾浓度分别按1∶1、1∶2和1∶3投加进行配比,每组做4次取各次测定石油类污染物浓度和pH的平均值作为实验值。
2 实验数据分析
2.1 石油类污染物氧化去除率
在弱碱性溶液中高铁酸钾氧化分解石油类污染物中链烃的化学反应方程式见式(2),其它芳烃类污染物的氧化反应类似。氧化反应产物小分子有机酸能被氧化为CO2,未被氧化的累积在溶液中。

2OH-1+(小分子有机酸)
(2)
在化学反应方程式(2)中,石油类污染物氧化分解产物小分子有机酸主要由甲酸、草酸、乙酸、丙酸和正丁酸组成。在小分子有机酸累积量计算中,考虑到石油类污染物氧化时间长,可忽略易于氧化的甲酸和草酸累积量。同时考虑到乙酸、丙酸和正丁酸为一级离解且离解常数在同一数量级,可采用乙酸,离解常数K=1.74×10-5等效计算溶液pH下降的小分子有机酸累积量。在石油类污染物氧化过程中,高铁酸钾始终保持在稀溶液状态,不考虑应因氧还原高铁酸钾释放的OH-1量。对各实验水样测试数据分析显示,理论计算值1∶3过量投加高铁酸钾的实验水样,石油类污染物能被稳定地氧化去除。各实验水样连续3次取样测得石油类污染物浓度和pH值趋于稳定的时间接近,平均时间25h为氧化反应结束。各水样测得的石油类污染物最大氧化去除率97%,平均氧化去除率为89.46%,氧化剩余石油类污染物平均浓度0.17mg/L。各水样中剩余石油类污染物浓度随氧化反应时间降低的趋势,见图1。

图1 剩余石油污染物类浓度随反应时间变化趋势图
由图1可知,各实验水样中石油类污染物氧化去除速率前期97%,后期降低为8.1%。初始浓度高的实验水样,前期氧化去除速率较大。在氧化反应进行15h后,各实验水样的氧化去除速率明显下降,25h后趋于稳定不再下降。氧化反应结束时,测得的4个实验水样中剩余石油类污染物浓度0.08~0.16 mg/L,平均浓度0.13mg/L,最大浓度0.23mg/L,最小浓度0.10mg/L。在氧化反应结束时,剩余石油类污染物浓度及其与初始浓度值的百分比,具体见表2。

表2 石油烃类有机物剩余浓度及其占初始浓度百分比
由表2分析可知,氧化反应结束时,水样浓度在5.02mg/L石油类污染物剩余量与初始量的比值最小。浓度为2.05 mg/L、1.01 mg/L、0.52 mg/L 3个水样,剩余石油类污染物量与初始量比较高,存在未被氧化分解的石油烃类污染物。氧化反应结束时各实验水样石油类污染物氧化去除率具体见表3。

表3 石油烃类污染物氧化去除率
2.2 小分子有机酸累积
实验测得各实验水样pH变化趋势见图2。在投加高铁酸钾氧化剂后,在快速混合离解初期释放出OH-1,pH在短时间内快速上升。实验过程随着石油类污染物的氧化和小分子有机酸累积pH下降至7.2。
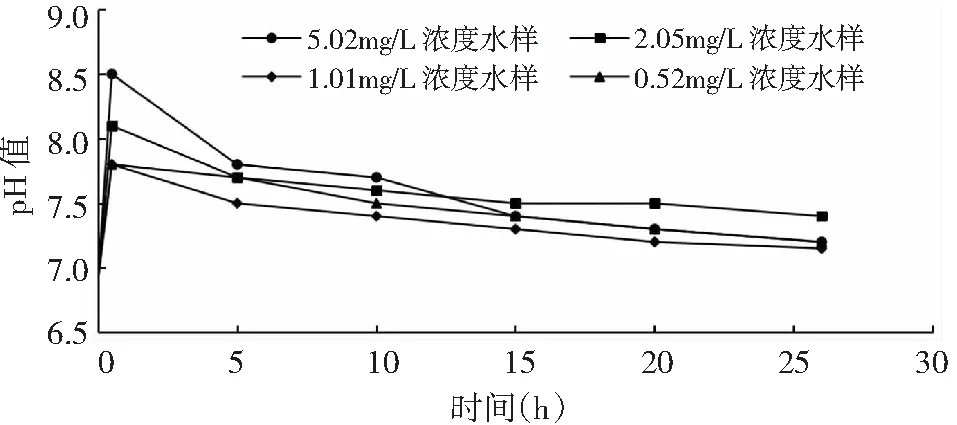
图2 各实验水样的pH变化趋势图
由图2中pH降低量和碳酸盐平衡关系式(3)~式(5)及乙酸离解方程式(6),可计算得到氧化反应结束时,小分子有机酸累积量,具体见表4。表中同时给出了按乙酸换算的小分子有机酸的COD浓度,及其与初始石油类污染物COD浓度比值。初始石油类污染物COD浓度根据0#轻质柴油的统计分子式换算得到。
[H+][OH-]=K1
(3)
(4)
(5)
(6)
式中,K1为水的离子积常数,K1=1×10-14;K2为碳酸一级电离常数,K2=4.4×10-7;K3为碳酸二级电离常数,K3=5.6×10-11。

表4 小分子有机酸累积浓度值
乙酸、丙酸和正丁酸三者摩尔质量接近,乙酸摩尔质量最小为60g/moL丙酸和正丁酸分别为74g/moL、88.11g/moL,表4中计算小分子有机酸累积浓度是一个下限值。由表4可知在氧化反应结束时,按乙酸等效计算得到的小分子有机酸COD占初始石油类污染物COD比值在2%~3.5%之间,与表4中5.02 mg/L的石油类污染物氧化去除率相比,石油类污染物氧化分解产生的小分子有机酸累积量为0.21 mg/L。
2.3 [Fe6+]过量状态
投加高铁酸钾后,混合离解时间短石油类污染物氧化量小,忽略初期因石油类污染物氧化消耗的[Fe6+]量。从化学反应方程式(1)可知,在高铁酸钾混合离解初期,被氧还原而释放的[OH-1]=0.5[Fe6+]。根据图2中测得的pH初期上升值和碳酸盐平衡关系式(3)~式(5),计算投加高铁酸钾后,混合离解初期[Fe6+]还原量具体见表5。
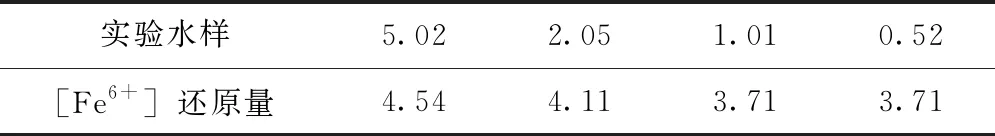
表5 高铁酸钾混合离解初期的[Fe6+]还原量
忽略氧化分解后转化为累积小分子有机酸的小量石油类污染物。根据图1中测定的石油类污染物剩余浓度,以及表5中混合离解初期[Fe6+]消耗量,采用0#轻质柴油统计分子式,计算得到在各反应时间,剩余[Fe6+]量与石油类污染物量的比值,具体见图3。
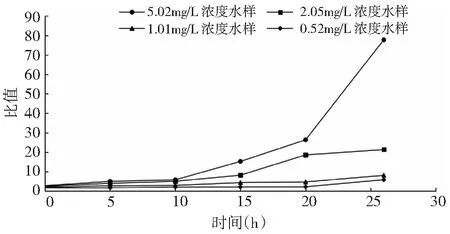
图3 剩余[Fe6+]量与剩余石油类污染物量比值变化趋势图
从图3可知,在石油类污染物的氧化分解过程中,[Fe6+]始终处于过量状态且随着氧化反应进行,剩余石油类污染物浓度降低,在[Fe6+]过量增加5.02mg/L浓度实验水样[Fe6+]过量比值高于其它3个低浓度的实验水样。在氧化反应结束时,其[Fe6+]过量比值达到70以上,有利于石油类污染物组分的氧化分解。
3 氧化反应动力学分析
在石油类污染物氧化分解过程中,[Fe6+]量始终保持过量状态忽略其对氧化反应速率的影响。石油类污染物氧化速率仅与其自身浓度相关。根据图1中测定的剩余石油类污染物浓度,经线性回归计算,可得到石油类污染物的氧化速率与其浓度的关系式(7),相关系数R=0.75,相对误差Er=5.8%。
(7)
式中:C-石油污染物浓度,mg/L。
用回归线性方程式(7)计算各反应时间石油类污染物剩余浓度与测定值吻合较好,具体见图1。表明在完全混合状态,高铁酸钾氧化剂过量的条件下,低浓度石油类污染物的氧化反应可近似为一级反应。
4 结 论
本文采用0#轻质柴油模拟石油类污染物,实验研究了石油类污染物浓度分别为5.02mg/L、2.05mg/L、1.01mg/L和0.52mg/L 的4个实验水样,在高铁酸钾氧化剂过量的条件下,石油类污染物的氧化去除规律。并对氧化产物小分子有机酸的累积量和高铁酸钾氧化剂的过量状态进行了计算分析。研究表明:
4.1 在pH=7.23弱碱性条件下,水样浓度分别为5.02mg/L、2.05mg/L、1.01mg/L和0.52mg/L时,石油类污染物的平均氧化去除率为89.46%,最大97.51%,氧化反应结束时间为25h。
4.2 在过量高铁酸钾存在条件下,低浓度石油类污染物的氧化反应可近似为一级反应。石油类污染物氧化分解产生的小分子有机酸累积量为0.21 mg/L,氧化反应结束后,溶液pH在7.2~7.4之间,保持在弱碱性状态。
4.3 按照理论计算值1∶3投加高铁酸钾量,能稳定地氧化低浓度石油类污染物。在石油类污染物氧化过程中[Fe6+]浓度始终处于较高的过量状态,有利于石油污染物组分氧化分解。
猜你喜欢
中国资源综合利用(2022年9期)2022-10-13
现代矿业(2022年3期)2022-04-09
商品与质量(2021年7期)2021-11-23
矿产综合利用(2020年1期)2020-07-24
当代水产(2019年11期)2019-12-23
中国调味品(2019年10期)2019-10-16
制造技术与机床(2019年9期)2019-09-10
探索科学(学术版)(2019年12期)2019-07-14
绿色科技(2018年24期)2019-01-19
中国茶叶加工(2015年3期)2015-02-27
