
CnAβ基因对大鼠RBL-2H3细胞脱颗粒的影响
2021-02-27 04:57李艳红吴正理
畜牧兽医学报 2021年2期
李艳红,刘 洁,吴正理
(西南大学水产学院 淡水鱼类资源与生殖发育教育部重点实验室,重庆 400715)
肥大细胞(mast cell, MC)是一种起源于骨髓或者其他造血组织的多功能造血干细胞,在组织中分化成熟、并能在组织中长期存活的一类炎症效应颗粒细胞。成熟的肥大细胞表达2种主要的表面受体:高亲和力的IgE受体(high affinity IgE receptor, FcεRI)和干细胞因子(stem cell factor, SCF)受体c-Kit(CD117)[1-2]。肥大细胞作为一类重要的免疫效应细胞,分布于各种组织、器官的表皮或黏膜上,在IgE介导的过敏性疾病中发挥核心作用[1]。通过膜上组成型表达的FcεRI结合并交联抗原依赖性的IgE,可致敏、活化肥大细胞,继而触发胞内信号级联反应,促进各种炎性介质的释放,包括其细胞质颗粒内的多种生物活性物质(如组胺),以及从头合成的细胞因子和趋化因子[3-4]。过敏疾病是肥大细胞炎症介质的过度产生和释放的一种表现[5],在人[6-8]和养殖动物[9-10]中发生的概率越来越大,而且养殖宠物和人之间可以相互影响产生过敏反应[9, 11]。随着养殖业的快速发展,食物源过敏事件[12-15]、疫苗过敏事件[16-17]频发,给养殖生产带来较大的经济损失。为此,深入了解动物过敏的分子机制,有利于预防和治疗动物过敏性疾病,为畜牧业的健康生产提供理论依据。
钙调磷酸酶(calcineurin, CaN)是一种参与多种细胞功能调节的多功能信号酶,由催化亚基(CnA)和调节亚基(CnB)组成的异源二聚体,在许多生物学过程以及疾病的发生中起关键作用,如在cAMP代谢、微管组装、骨骼肌代谢、血管平滑肌细胞、T细胞的活性(活化、分化及增殖)等过程中起至关重要的作用[18];在心肥大、骨质疏松、骨骼肌纤维化、变态反应和自身免疫性疾病[19-20]及神经元生长和突触可塑性[21]等多种疾病的免疫应答中扮演中心角色。CaN-NFAT信号通路可通过活化T细胞而参与哮喘的整个发病过程[22-23],同时,参与了控制免疫、神经、心血管、骨骼系统发育和功能的信号级联放大调节[20];钙调磷酸酶还可以通过激活NFκB信号途径刺激天然免疫反应[24];且受钙调磷酸酶调节的转录因子NFAT[25]和NFκB[26]均参与肥大细胞的脱颗粒过程,但钙调磷酸酶是否参与肥大细胞颗粒的形成以及脱颗粒过程,仍未见报道。本研究旨在探究钙调磷酸酶对肥大细胞脱颗粒的影响,以期为动物过敏性疾病的预防提供理论基础。
1 材料与方法
1.1 药品与试剂
胎牛血清、培养基(DMEM/F12、Opti-MEM)、磷酸缓冲液(PBS)、双抗、胰酶均购自美国Gibco公司。限制性内切酶、嘌呤霉素、TaqDNA聚合酶、T4 DNA 连接酶、BpiⅠ内切酶、Lipofectamine 3000、DNA提取试剂盒、反转录试剂盒、Trizol试剂购自Invitrogen公司。A23187、Carnoy固定液、HBSS缓冲液、LPS、anti-DNP IgE、DNP-BSA、甲苯胺蓝、C48/80、p-NAG、NP-40、WST-1购自Sigma公司。pX459质粒为广西大学林文珍教授馈赠。
1.2 细胞培养
大鼠肥大细胞系(RBL-2H3,中国科学院昆明动物研究所)培养于37 ℃,5% CO2细胞培养箱中,贴壁生长,当细胞覆盖率达80%以上时以1∶3传代,在细胞对数生长期进行试验。
1.3 pX459-CnAβ-sgRNA载体构建与检测
使用在线软件CRISPR direct在大鼠CnAβ基因序列(NC_005114.4)的第一个外显子上设计3个sgRNA寡核苷酸序列。根据质粒pX459中BpiⅠ酶切位点的结构,在sgRNA正义链寡核苷酸序列的5′端添加“CACC”,并在反义链寡核苷酸序列的5′端添加“AAAC”。为提高转录效率,若sgRNA正义链寡核苷酸序列的“CACC”后面的碱基不是“G”,则需要添加碱基“G”或“GG”,并在反义链寡核苷酸序列的3′端添加碱基“C”或“CC”,同时,设计无义DNA序列作为对照(control oligo),序列设计结果见表1。将合成的两条单链sgRNA退火形成双链sgRNA,然后,经BpiⅠ酶切再与经BpiⅠ酶切的pX459载体连接,转化入大肠杆菌DH5α,最后,进行阳性质粒筛选和测序验证。
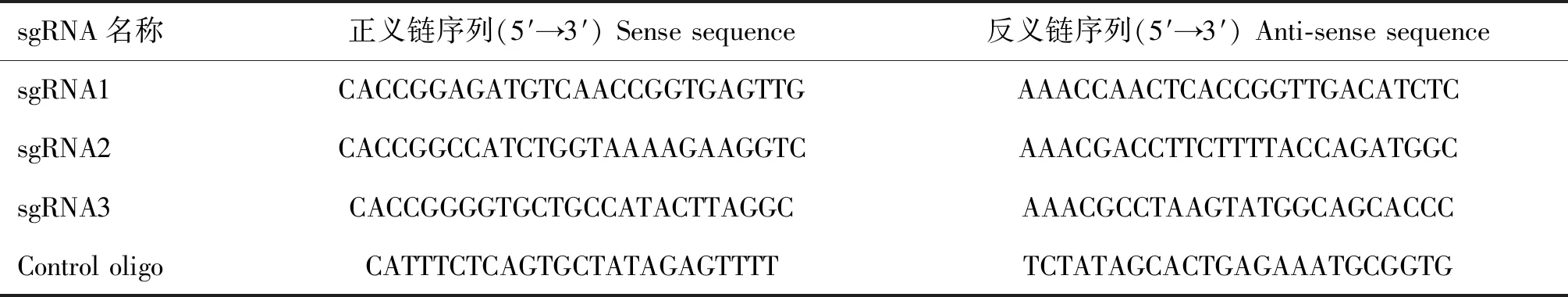
表1 CnAβ基因的sgRNA序列
1.4 CnAβ基因敲除RBL-2H3细胞系的构建
将重组质粒或无义DNA对照质粒用LipofectamineTM3000转染入大鼠RBL-2H3细胞,用终浓度1 μg·mL-1嘌呤霉素对转染细胞进行筛选以构建稳定的CnAβ基因敲除细胞系。具体操作:取对数生长期的RBL-2H3细胞,以5×105cells·孔-1均匀铺于6孔细胞培养板中,培养24 h;用Opti-MEMTM培养基分别稀释脂质体LipofectamineTM3000和3种等量混合的pX459-CnAβ-sgRNA质粒(敲除组/knockout,KO)或无义DNA对照pX459质粒(MOCK)后按1∶1的比例混匀,室温静置15 min。将配制好的“质粒-脂质体”混合物缓慢加入细胞培养板中,边加边摇匀。培养24 h后,去上清,每孔加入2 mL含1 μg·mL-1嘌呤霉素的完全培养基;72 h后,更换含1 μg·mL-1嘌呤霉素的完全培养基;待培养96 h后,观察细胞存活量,并稀释成单细胞扩大培养。最后,提取细胞DNA,以CnAβ基因引物(上游引物:5′-ACATGTAGGATGACTGGGTT-3′;下游引物:5′-AAGAGCAAAGGAAGGTGTTG-3′)进行PCR扩增,并对PCR扩增产物进行测序验证,以筛选CnAβ基因敲除的RBL-2H3细胞株。
1.5 RT-PCR验证CnAβ基因的表达
用Trizol试剂提取细胞RNA,并反转录成cDNA,以GAPDH基因为内参,分别对CnAβ基因和GAPDH基因进行PCR扩增,扩增产物经琼脂糖凝胶电泳检测基因的表达水平。RT-PCR扩增引物分别为CnAβ基因(上游引物:5′-ATACTTAGGCGGGAGAAAACC-3′,下游引物:5′-CCATACAAGCTTCATAGACC-3′),GAPDH基因(上游引物:5′-ACTCCCTCAAGATTGTCAGC-3′,下游引物:5′-ACATTGGGGGTAGGAACACG-3′)。
1.6 WST-1染色测定细胞增殖能力
取对数生长期的细胞,分别稀释成0.5×105、1.0×105,或5.0×105cells·mL-1,以100 μL·孔-1接种到96孔细胞培养板中,每个浓度设置3个重复,在405 nm处测定吸光值,此时记为0 h。然后每孔分别加入10 μL WST-1,混匀,于5% CO2培养箱中继续培养,分别于24、48、72和96 h测定吸光值(A405 nm)。
1.7 甲苯胺蓝染色检测
在24孔细胞培养板中加入灭菌盖玻片,取对数生长期细胞加入培养孔中(2×105cells·孔-1),在37 ℃、5% CO2培养箱中过夜培养,制作细胞爬片。弃上清,PBS漂洗细胞后,加入含10 μmol·L-1A23187的新鲜培养基刺激1.5 h。弃上清,PBS漂洗细胞,取出细胞爬片,用Carnoy液固定10 min,66%乙醇脱水10 min,0.5%醋酸处理1 min。加入0.5%甲苯胺蓝染色1.5 h,蒸馏水洗涤3次,室温干燥。用中性树脂封片,于显微镜(40×)下观察。
1.8 DNP、C48/80和LPS诱导细胞脱颗粒
取对数生长期的细胞(用于DNP-BSA刺激的细胞需用1 μg·mL-1anti-DNP IgE致敏过夜),用HBSS缓冲液重悬计数,调整细胞密度为1×106cells·mL-1。将细胞以每孔100 μL接种于96孔 板细胞培养板中。用HBSS缓冲液稀释DNP-BSA、LPS和C48/80,使其终浓度分别为DNP-BSA(0.00、0.01、0.10、1.00、10.00、50.00、100.00 μg·mL-1),LPS(0.0、0.1、1.0、10.0、100.0 μg·mL-1),C48/80(0、10、20、40、60、80、100、200 μg·mL-1)。每孔分别加入100 μL稀释后的DNP-BSA,LPS,C48/80,空白对照加入100 μL HBSS缓冲液。置于37 ℃,5% CO2培养箱中孵育1.5 h后,于1 500 r·min-14 ℃离心10 min,取上清(细胞培养上清液)。加入200 μL 1% NP-40细胞裂解液于细胞沉淀中,重悬细胞,室温静置10 min后,3 400 r·min-14 ℃离心5 min,取上清(细胞裂解上清液)。细胞脱颗粒效率采用β-氨 基己糖苷酶(β-hexosaminidase)的释放率来表示,分别取50 μL细胞培养上清液、细胞裂解上清液或HBSS缓冲液(对照)于96-孔细胞培养板中,每孔加入50 μL 1 mmol·L-1p-NAG液,37 ℃孵育2 h;加入200 μL 0.1 mol·L-1碳酸盐缓冲液终止反应,在酶标仪中测定吸光值(A405 nm)。按照如下公式计算细胞脱颗粒效率。
脱颗粒%=(A405 nm 细胞培养上清液-A405 nm 对照)/(A405 nm 细胞培养上清液+A405 nm 细胞裂解上清液-2 A405 nm 对照)×100。
1.9 统计学分析
试验结果以“平均数±标准误”表示,试验重复3次。试验数据采用t检验,*.P<0.05 认为差异有统计学意义。
2 结 果
2.1 pX459-CnAβ-sgRNA载体构建
将成功转化的候选单菌落大肠杆菌DH5α细胞提取质粒,然后用BpiⅠ 酶切候选重组质粒pX459-CnAβ-sgRNA(sgRNA1、sgRNA2、sgRNA3)并进行电泳检测(图1A)。由于外源片段的插入破坏了质粒BpiⅠ 的酶切位点,因此,不能被BpiⅠ 酶切的质粒即插入了外源DNA片段;而能被BpiⅠ 酶切的质粒则表示未插入外源DNA片段,如图1A 中sgRNA1-P1、sgRNA3-P4。最后,挑取3个质粒(sgRNA1-P5、sgRNA2-P2和sgRNA3-P3)进行测序验证(图1B),结果表明,这3种sgRNA均成功插入pX459质粒,可用于后续CnAβ基因敲除RBL-2H3细胞株的构建。
2.2 CnAβ基因敲除RBL-2H3细胞系的构建
将成功构建的pX459-CnAβ-sgRNA质粒载体用脂质体3000转入RBL-2H3细胞中,经嘌呤霉素筛选出49个单克隆细胞株,通过DNA测序鉴定后显示其中12个单克隆细胞株均有一定程度的敲除(图2A),其敲除效率为24.5%。笔者选择敲除最多的6号克隆株(KO)作为进一步研究对象,提取该细胞株基因组DNA,经PCR扩增后显示,与野生型(WT)和无义DNA对照(MOCK)组细胞相比,KO组细胞的CnAβ基因片段明显变短(图2B),测序结果显示,已成功敲除整个外显子1及附近的381 bp序列片段(图3)。为进一步了解该片段的缺失是否影响CnAβmRNA的表达,提取细胞总RNA,并反转录cDNA后进行RT-PCR扩增验证,结果显示,DNA片段缺失后CnAβ基因不能正常表达(图4),而野生型细胞(WT)和无义DNA对照细胞(MOCK)均正常表达,且经钙离子载体A23187刺激前后其表达量无明显变化,表明CnAβ基因为组成性表达,CnAβ基因敲除细胞系(KO)构建成功。

图3 敲除细胞与野生细胞DNA序列比对
WT.野生细胞;KO.CnAβ基因敲除细胞; MOCK.无义DNA转染细胞;0、1、3.A23187 刺激时间(h)
2.3 CnAβ基因对RBL-2H3生长的影响
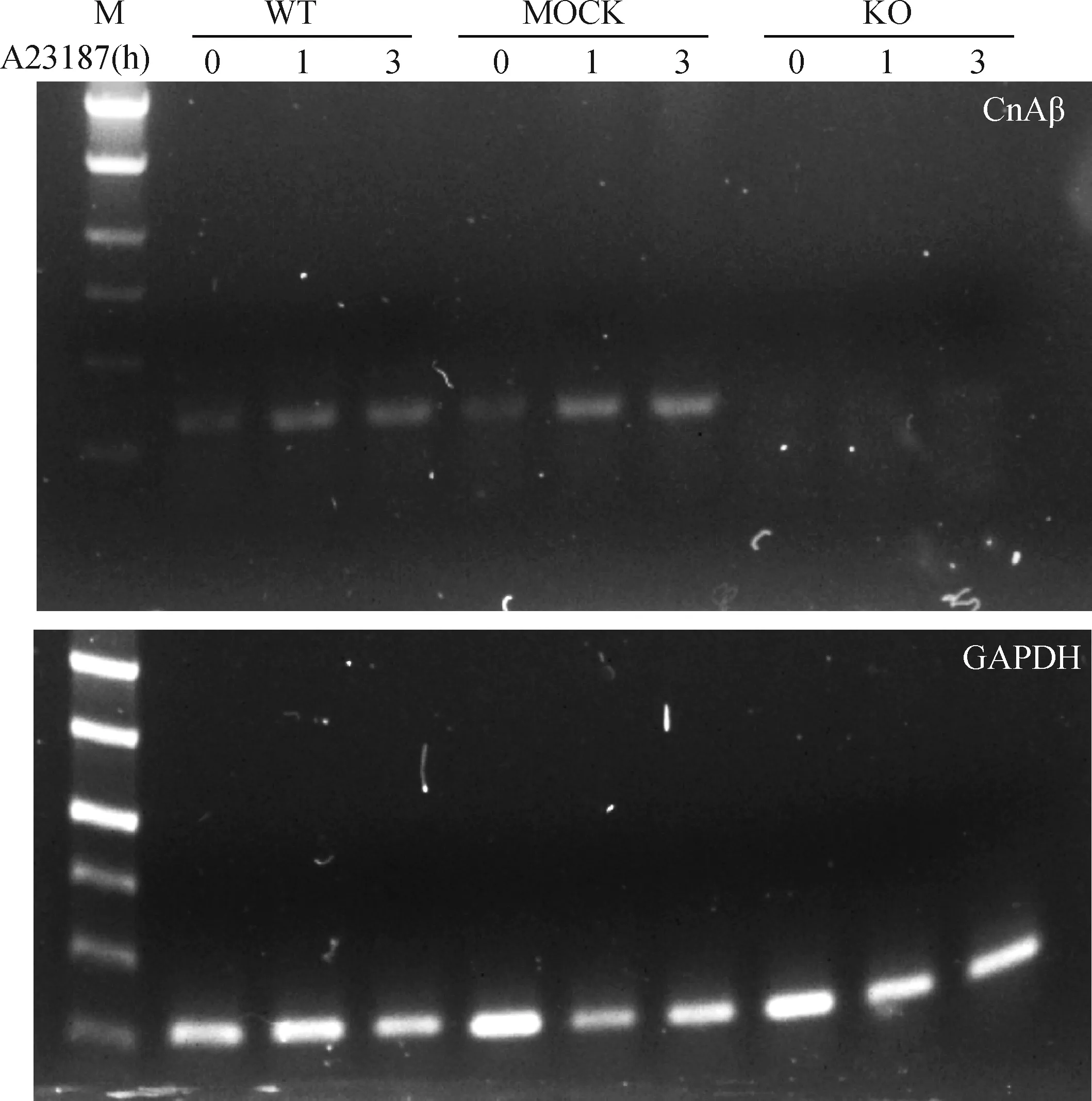
为了研究CnAβ基因缺失后是否影响RBL-2H3细胞的生长,采用WST-1检测细胞的增殖。结果如图5所示,3种细胞以不同浓度(0.5×105、1.0×105或5.0×105cells·mL-1)接种后,细胞增殖均无显著性差异,在24 h内均可到达生长平稳期。结果表明,敲除CnAβ基因对RBL-2H3细胞生长无影响。
A.0.5×105 cells·mL-1;B.1.0×105 cells·mL-1;C.5.0×105 cells·mL-1; WT.野生细胞;KO.CnAβ基因敲除细胞; MOCK.无义DNA转染细胞
2.4 CnAβ基因对RBL-2H3细胞颗粒含量的影响
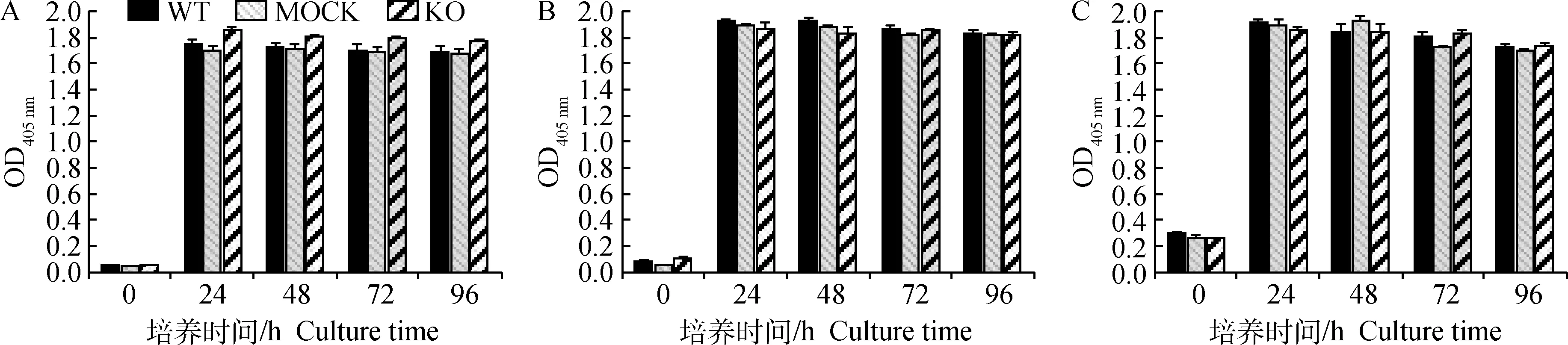
为了研究CnAβ基因缺失对RBL-2H3细胞内颗粒形成的影响,本试验首先通过制作细胞爬片,利用钙离子载体A23187刺激细胞1.5 h后经甲苯胺蓝试剂染色观察(图6)。结果显示,3种细胞在静息状态下均较饱满,边缘整齐,染色较深,且3种细胞没有明显差异;当细胞脱颗粒后,细胞变大、变圆,细胞膜边缘不整齐,细胞质变得稀疏,染色变浅,并出现很多空泡结构,但3种细胞结构无明显差异。进一步检测经A23187刺激后细胞的β-氨基己糖苷酶(β-hexosaminidase)释放率(图7),结果显示,3种细胞β-氨基己糖苷酶释放率无显著差异。上述结果表明,CnAβ基因缺失不影响RBL-2H3细胞颗粒的形成和颗粒的含量。
图6 A23187诱导前后3种细胞的形态(40×)
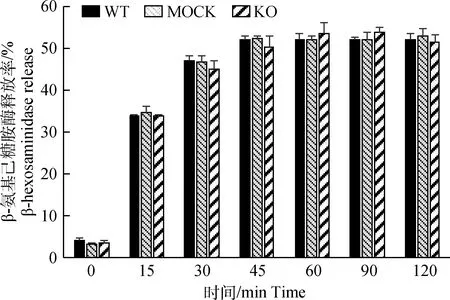
图7 10 μmol·L-1 A23187刺激细胞脱颗粒
2.5 CnAβ基因对RBL-2H3细胞脱颗粒的影响
由于A23187是钙离子载体,其作用原理是在增加细胞内的钙离子浓度从而激活细胞,而不是通过细胞表面的受体信号调节。因此,为了进一步研究CnAβ基因对RBL-2H3细胞表面受体调节细胞脱颗粒的影响,笔者选择了C48/80、IgE/DNP-BSA和LPS作为刺激物检测细胞表面受体信号调节细胞脱颗粒的影响。结果显示,C48/80、IgE/DNP-BSA和LPS刺激RBL-2H3细胞脱颗粒效应均存在剂量效应(图8A、C、E),且C48/80的最佳刺激浓度为100 μg·mL-1(图8A),DNP-BSA的最佳刺激浓度为10.00 μg·mL-1(图8C),LPS的最佳刺激浓度为1.0 μg·mL-1(图8E)。LPS刺激RBL-2H3细胞脱颗粒效应显著低于C48/80和IgE/DNP-BSA刺激。3种刺激因子分别刺激3种细胞,野生型细胞(WT)和无义DNA突变细胞(MOCK)脱颗粒效应无显著差异,但均显著高于CnAβ基因缺失细胞(KO)的脱颗粒效应。进一步检测了3种刺激因子在最佳刺激浓度下的刺激时间效应,发现3种刺激因子均存在刺激时间依赖性,刺激60 min后,细胞脱颗粒效应均达到峰值(图8B、D、F);与剂量效应类似,野生型细胞(WT)和无义DNA突变细胞(MOCK)脱颗粒效应无显著差异,但均显著高于CnAβ基因缺失细胞(KO)的脱颗粒效应。综上表明,CnAβ基因缺失显著抑制了RBL-2H3细胞表面受体信号介导的细胞脱颗粒效应。
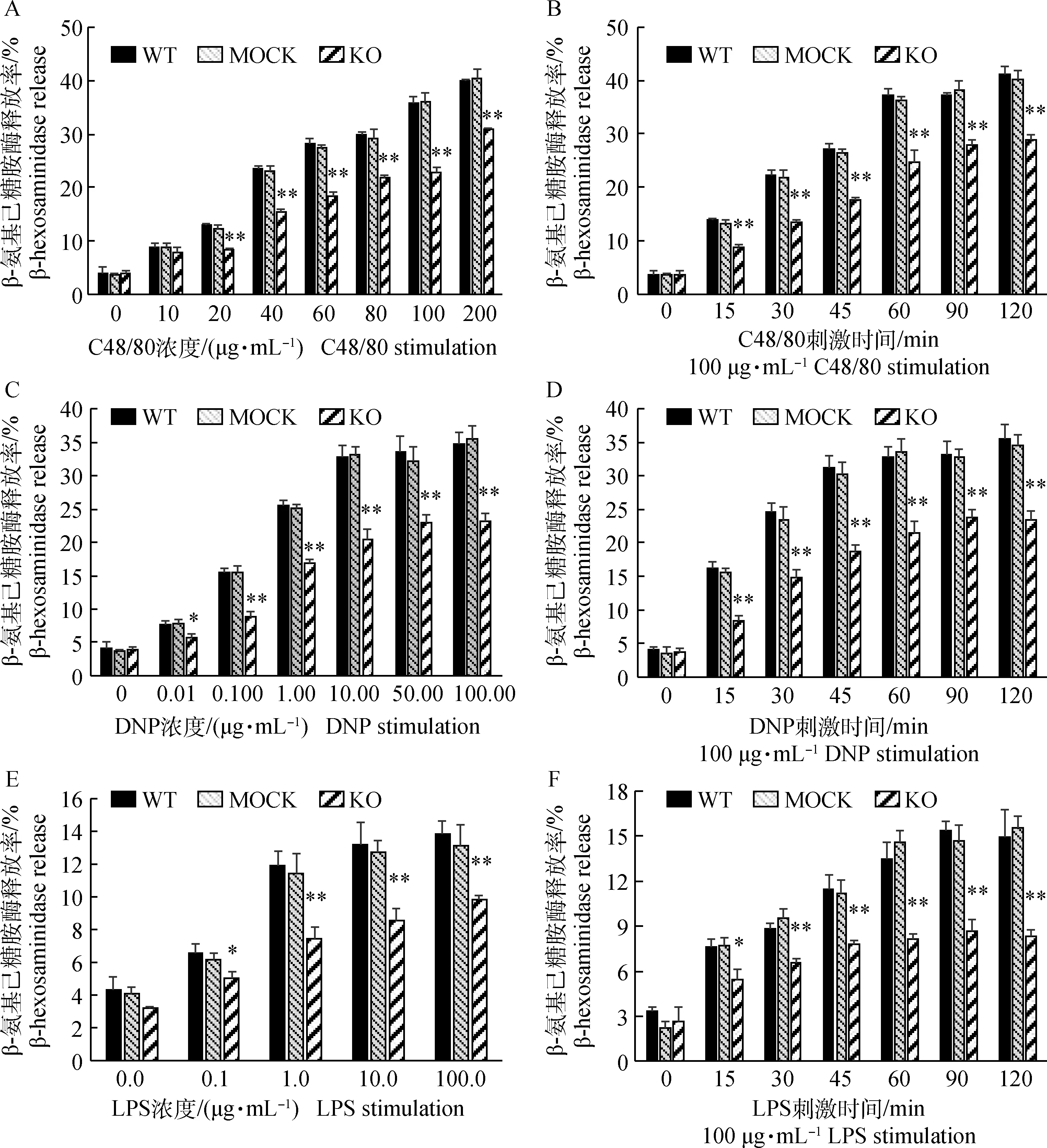
A.C48/80刺激细胞脱颗粒的浓度效应; B.100 μg·mL-1 C48/80刺激细胞脱颗粒的时间效应; C.DNP刺激细胞脱颗粒的浓度效应; D.10 μg·mL-1 DNP刺激细胞脱颗粒的时间效应; E.LPS刺激细胞脱颗粒的浓度效应; F.1 μg·mL-1 LPS刺激细胞脱颗粒的时间效应.*.P<0.05; **.P<0.01
3 讨 论
本研究成功构建了CnAβ基因缺失的RBL-2H3细胞株,CnAβ基因的缺失并不影响RBL-2H3细胞的生长繁殖、细胞颗粒的形成和颗粒的含量,但显著抑制了细胞表面受体介导的细胞脱颗粒效应。这表明,CnAβ基因在细胞表面受体介导的肥大细胞脱颗粒效应的信号通路中起着关键作用,为肥大细胞脱颗粒的分子机制研究提供了研究基础,同时,为过敏性疾病的防治提供了研究靶点。
利用CRISPR/Cas9质粒构建基因缺失型细胞株已经被广泛应用[27],但应用在免疫细胞的基因突变还比较少。本文利用CRISPR/Cas9质粒对RBL-2H3细胞进行CnAβ基因的敲除,但敲除效率较低,仅24.5%。这可能是由于免疫细胞具有自我保护机制,外源物质在细胞内更容易被降解。
钙调磷酸酶是一种参与多种细胞功能调节的多功能信号酶,主要通过调节转录因子NFAT和NF-κB的活性参与到细胞信号转导过程[20]。但钙调磷酸酶是否参与肥大细胞脱颗粒过程并不清楚。本研究结果说明,CnAβ基因不参与肥大细胞颗粒的形成,但广泛参与由细胞表面受体介导的肥大细胞脱颗粒过程。肥大细胞激活有IgE依赖性和IgE非依赖性两种模式。IgE通过与其受体FcεRI特异性结合发挥作用,是肥大细胞最常见的一种激活方式,同时也是IgE介导过敏反应的基础[28]。C48/80(Compound 48/80)是N-甲基-对甲氧基苯乙胺和甲醛缩合产生的多聚体混合物,可激发Gi-蛋白依赖的肥大细胞脱颗粒,有效地活化肥大细胞[29]。研究表明,对大鼠进行腹腔注射C48/80或直接刺激RBL-2H3细胞可引起大鼠肥大细胞脱颗粒释放组胺和β-氨基己糖苷酶[30-31]。脂多糖(LPS)是一种存在于革兰阴性菌胞壁的多糖,研究发现,LPS可通过结合细胞表面受体TLR(toll like receptor)免疫刺激后可诱导肥大细胞脱颗粒,释放组胺和β-氨基己糖苷酶,以及炎性因子IL-1、1L-6和TNF-α等[5, 32]。β-氨基己糖苷酶是肥大细胞或嗜碱性粒细胞脱颗粒后释放的一种预合成物质。只有机体过敏时才会引发血浆中β-氨基己糖苷酶的含量明显升高,可以作为肥大细胞脱颗粒现象的标志物[33]。本研究通过检查β-氨基己糖苷酶的释放率,证明了CnAβ基因参与调控FcεRI、Gi-蛋白和TLR介导的肥大细胞脱颗粒过程,也表明CnAβ基因在一定程度上调节了机体的免疫反应。
4 结 论
成功构建了CnAβ基因敲除RBL-2H3细胞系,CnAβ基因缺失不影响肥大细胞的生长、细胞内颗粒的形成和颗粒的含量,但CnAβ基因缺失严重抑制了经细胞表面受体信号诱导的肥大细胞脱颗粒效应,表明CnAβ基因参与调节肥大细胞的脱颗粒过程。
猜你喜欢
生物技术通讯(2019年1期)2019-02-18
中华临床免疫和变态反应杂志(2018年6期)2019-01-17
天然产物研究与开发(2018年9期)2018-10-08
中成药(2018年5期)2018-06-06
中成药(2017年8期)2017-11-22
中国科技纵横(2016年20期)2016-12-28
中国医药科学(2015年15期)2015-02-27
天然产物研究与开发(2014年3期)2014-04-27
重庆医学(2014年9期)2014-03-27
医学研究与教育(2014年2期)2014-03-11
