
浅析如何推进2020年版《中国药典》在制药企业的实施工作
2021-02-27 05:18北京协和药厂102600王涛何丽娟
首都食品与医药 2021年3期
北京协和药厂(102600)王涛 何丽娟
2020年版《中华人民共和国药典》(以下简称《中国药典》)于2020年12月30日实施,其颁布实施,将有利于整体提升我国药品标准水平,进一步保障公众用药安全[1]。
国家药品标准是国家为保证药品质量,对药品的质量指标、检验方法和生产工艺等作出的强制性规定,是药品生产、流通、使用和监管所必须遵循的法定技术要求。2019年12月1日实施的《中华人民共和国药品管理法》(以下简称《药品管理法》)中第二十八条明确规定:“药品应当符合国家药品标准。以国务院药品监督管理部门颁布的《中华人民共和国药典》和药品标准为国家药品标准。”
《中国药典》是国家药品标准体系的核心,关系到人民群众的身体健康和用药安全。《药品管理法》进一步明确了《中国药典》的法律地位,为药品研制、生产、经营、使用和监督管理活动提供了国家标准依据。制药企业要做好2020年版《中国药典》颁布实施和贯彻执行,确保对2020年版《中国药典》理解到位、执行到位、监督到位。
12020年版《中国药典》收载情况简述
2020年版《中国药典》[2]收载品种5911种,新增品种319种,修订3177种,不再收载10种,因品种合并减少6种。一部中药收载2711种,其中新增117种、修订452种。二部化学药收载2712种,其中新增117种、修订2387种。三部生物制品收载153种,其中新增20种、修订126种;新增生物制品通则2个、总论4个。四部收载通用技术要求361个,其中制剂通则38个(修订35个)、检测方法及其他通则281个(新增35个、修订51个)、指导原则42个(新增12个、修订12个);药用辅料收载335种,其中新增65种、修订212种。
《中国药典》自1953年第一版颁布实施以来,到2020年第十一版,《中国药典》的收载品种已从531种增至5911种。
2020年版《中国药典》四部的变化尤为突出(见附表),相比于2015年版《中国药典》四部,修订了92.1%的制剂通则,20.5%的检测方法及其他通则,40%的指导原则,78.5%的辅料品种。由于《药品注册管理办法》中第十四条明确规定:国家药品监督管理局建立化学原料药、辅料及直接接触药品的包装材料和容器关联审评审批制度。在审批药品制剂时,对化学原料药一并审评审批,对相关辅料、直接接触药品的包装材料和容器一并审评。故2020年版《中国药典》四部进行了较大幅度的修订,尤其是制剂通则和药用辅料。制剂通则更加体现制剂的全过程控制以及药用辅料的功能性。
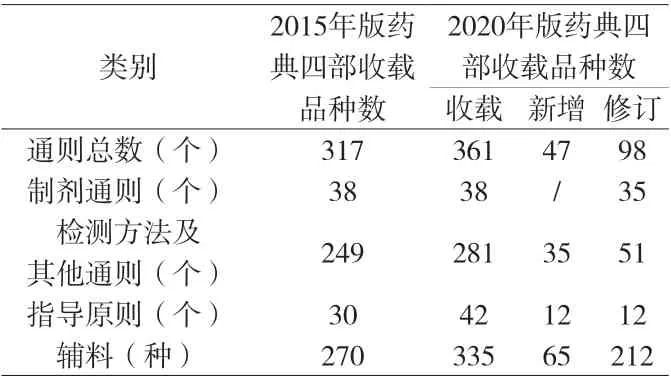
附表 2015年及2020年版《中国药典》四部收载情况比较
2020年版《中国药典》在品种收载、贯彻药品全生命周期管理理念、完善药品标准体系、加强药品安全性有效性控制、扩大成熟分析检测技术的应用、加强与国际药品标准协调等方面均取得了新的进展。
2 绘制思维导图,理清工作思路
2020年版《中国药典》正式实施前,制药企业需要做好充足准备,这项工作要以实验室为主导,QA、采购、仓储、生产、研发等多部门配合方可有效完成。首先,由实验室根据2020年版《中国药典》的收载内容,理清思路,明确2020年版《中国药典》正式实施后可能会对制药企业产生哪些方面的影响。对于产生的影响,制药企业应该采用哪些措施应对。梳理根据2020年版《中国药典》对制药企业的影响及应对措施,可以通过思维导图的方式展现。其次,根据思维导图的内容推进具体工作,实验室负责对比2015年及2020年版《中国药典》的异同点。然后对初步筛查的数据进行分类汇总,并且汇同相关部门开展评估分析工作。最后,在评估分析报告的基础上进入变更程序,开展变更工作,并完成分析方法确认、委托检验、物料的再检验、印刷性包材的修订、文件及记录的修订等具体工作。根据2020年版《中国药典》的推进工作以及修订内容,开展人员培训工作。
思维导图可以更加直观清晰地展现思路,以本企业的思维导图举例,具体分为初步筛查(见附图1),评估汇总(见附图2),汇总分析(见附图3)。
3 推进2020年版《中国药典》全面实施
3.1 初步筛查 根据绘制的思维导图,逐步完成2020年版《中国药典》在制药企业的推进工作。实验室首先开展2015年及2020年版《中国药典》的比对工作,比对两版药典的异同点并进行记录。比对的内容包括:凡例、通则、指导原则、品种正文等药典收载内容,并按照凡例修订、通则修订、指导原则修订、品名修订、贮藏修订、各品种检验项目修订、各品种检验限度修订等类别分类进行汇总。
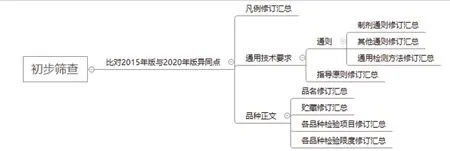
附图1 初步筛查思维导图
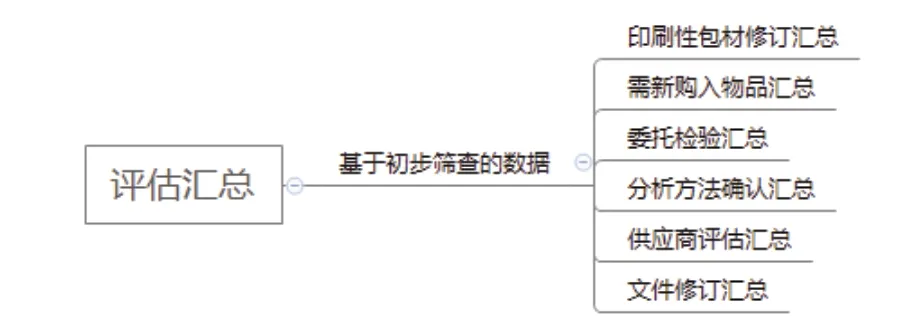
附图2 评估汇总思维导图
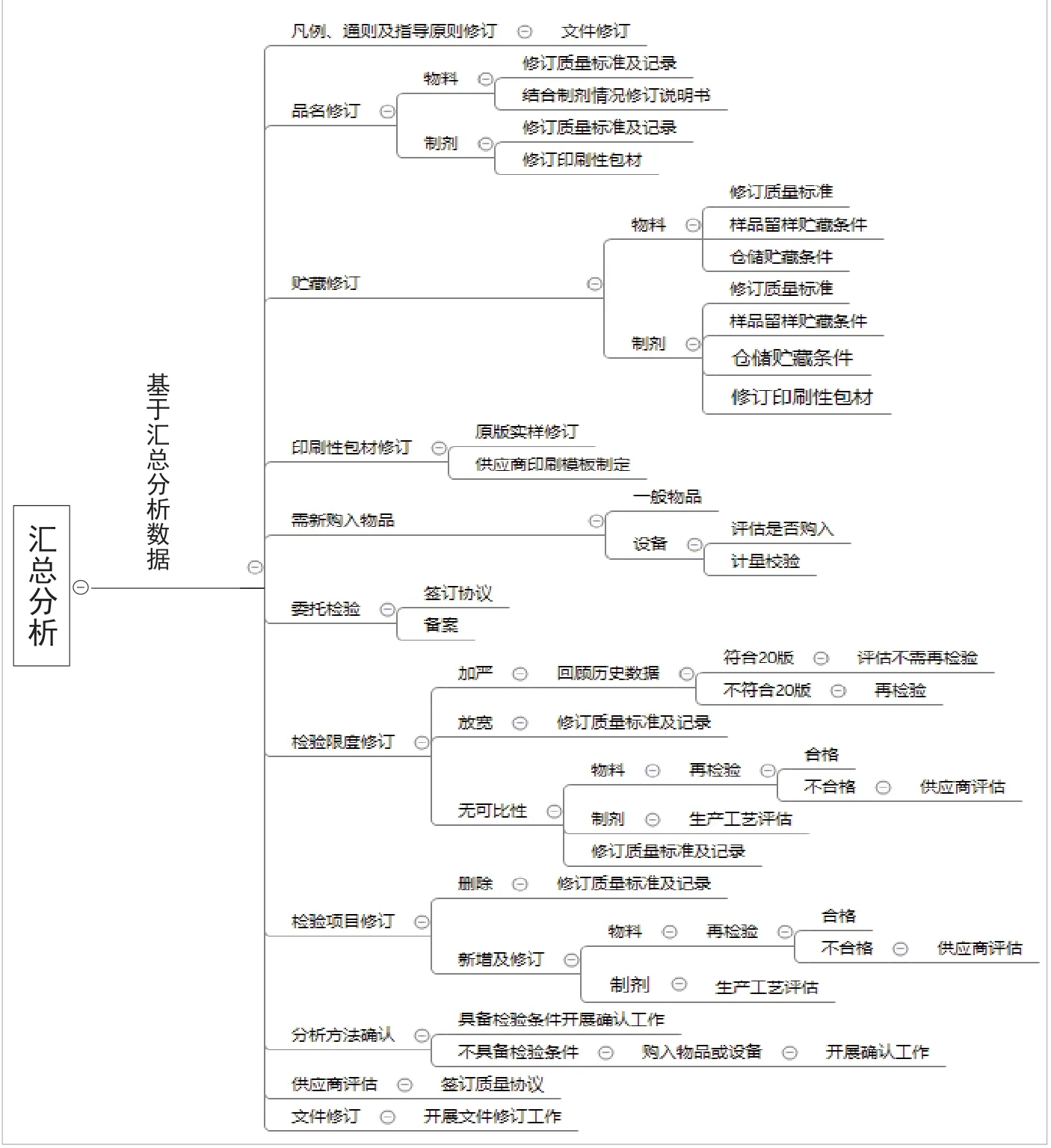
附图3 汇总分析思维导图
3.2 评估汇总 根据第二步收集的初步筛查的数据以及汇总的信息,筛选出印刷性包材修订、需购入物品、委托检验、分析方法确认、文件修订等内容再次进行汇总。
3.3 汇总分析
3.3.1 凡例修订的评估 凡例的修订会对实验室的基本操作产生影响,故需要修订与之相关的文件。凡例是为正确使用《中国药典》,对品种正文、通用技术要求以及药品质量检验和检定中有关共性问题的统一规定和基本要求。
3.3.2 通则修订的评估 通用检测方法的修订主要影响实验室检验工作,要考虑其修订对于标准操作规程的影响,同时考虑实验设备的更新以及检验员技术水平的提高。通则主要包括:制剂通则、其他通则、通用检测方法。制剂通则系为按照药物剂型分类,针对剂型特点所规定的基本技术要求。制剂通则的修订主要会对药品的生产、检验、包装与贮藏、运输等方面产生影响。生产部、QA部、仓储部均需考虑修订对其造成的影响。通用检测方法系为各品种进行相同项目检验时所应采用的统一规定的设备、程序、方法及限度等。
3.3.3 指导原则修订的评估 指导原则修订主要影响实验室的管理工作,包括稳定性的管理,分析方法验证、确认与转移,微生物实验室管理等。根据指导原则的修订内容修订相应管理文件。指导原则系为规范药典执行,指导药品标准制定和修订,提高药品质量控制水平所规定的非强制性、推荐性技术要求。
3.3.4 品名修订的评估 品名修订主要影响质量标准、检验记录以及说明书的修订。如物料的品名修订,首先需要修订其质量标准及检验记录,其次需要结合其相应制剂,判定是否需要修订其说明书、标签等印刷性包装材料的内容。如制剂的品名修订,首先需要修订其质量标准及检验记录,其次需要修订其说明书、标签等印刷性包装材料的内容。
3.3.5 贮藏修订的评估 贮藏修订主要影响质量标准、检验记录以及说明书的修订。如物料的贮藏修订,首先需要修订其质量标准,其次需要修订其样品及留样的管理规程,并通知仓储部门变更物料的贮藏条件。如制剂的贮藏修订,首先需要修订其质量标准,其次需要修订其样品及留样的管理规程,再次需要修订其说明书、标签等印刷性包装材料的内容,然后评估现有制剂的稳定性考察条件是否符合2020年版《中国药典》要求,并通知仓储部门变更制剂的贮藏条件。在制定物料及制剂的贮藏条件时,还需考虑安全要求,对于易燃易爆品,要兼顾药典和安全要求制定贮藏条件。
3.3.6 各品种检验限度修订的评估 检验限度修订主要影响分析方法学确认、物料及制剂的管理。2020年版《中国药典》与现行标准进行比对,各品种检验限度分为限度加严、放宽和无可比性三类。当限度放宽时,只需修订相应质量标准及检验记录。当限度加严时,还需对以往检验数据进行回顾,如以往检验数据符合2020年版《中国药典》的要求,则不需要对该物料按照新版药典进行检验。如以往检验数据不符合2020年版《中国药典》的要求,则需要对该物料按照新版药典进行检验,同时还要进行供应商的评价工作,按照新版药典的要求签订质量协议。当限度无可比性时,一般指检验方法发生变化。物料则需要对该物料按照新版药典进行检验,同时还要进行供应商的评价工作,按照新版药典的要求签订质量协议。若制剂的检验限度修订,还需开展生产工艺评估。
3.3.7 各品种检验项目修订的评估 检验项目修订主要影响分析方法学确认、物料及制剂的管理。2020年版《中国药典》与现行标准进行比对,各品种检验项目分为新增、修订和删除三类。对于删除的检验项目只需修订相应质量标准及检验记录。对于新增和修订的检验项目,还需评估是否需对在库物料及制剂进行质量评估。如制剂不符合2020年版《中国药典》要求,还需汇同生产和QA开展生产工艺评估与验证。如物料不符合2020年版《中国药典》要求,还需汇同采购部与QA部等开展供应商评估,重新签订质量协议或新增供应商。对于修订的检验项目,需要对其内容进行评估,如只是规范检验操作,对检验结果无影响,则无需再检验。若修订项目为物料的安全性指标,例如重金属、铁盐等,必须再检验。如物料的修订项目会对制剂质量造成影响,也需再检验,以满足制剂工艺要求,保证制剂成品的质量可靠。
3.3.8 印刷性包材修订的评估 根据印刷性包材修订的汇总内容,QA部需要开展原版实样的修订工作,除执行标准变更外,还需对变更内容进行备案。采购部门需要与印刷供应商重新制定印刷模板。印刷性包材的修订还需符合《药品生产质量管理规范》(2010年修订)[3]中第六章物料与产品第四节包装材料的相关规定。
3.3.9 需新购入物品的评估 需新购入物品包括设备、色谱柱、对照品、试药、试剂等。需要对需新购入设备进行评估,如设备价值较高,使用频次较低,可以评估是否可以委托检验该检验项目。新购入的设备需要符合《药品生产质量管理规范》(2010年修订)中第五章设备的相关规定。3.3.10 委托检验的评估 委托检验的物料及制剂的检验项目,因目前实验室不具备检验条件,经评估后可以委托检验。委托检验需要符合《药品生产质量管理规范》(2010年修订)中第十一章委托生产与委托检验的相关规定。
3.3.11 分析方法确认的评估 对于需要进行分析方法确认的物料及制剂进行评估,若具备检验条件则开展确认工作,若不具备检验条件,则需购入物品或设备后,待检验条件具备后开展确认工作。分析方法的确认还需符合《药品生产质量管理规范》(2010年修订)中第七章的确认与验证的相关规定。
3.3.12 供应商评估 质量管理部根据2020年版《中国药典》品种正文的修订内容,并结合品名修订评估、贮藏修订评估、印刷性包材修订评估、需新购入物品汇总、各品种检验项目修订评估、各品种检验限度修订评估、分析方法确认评估等内容,对供应商进行评估,签订质量协议。供应商评估还需符合《药品生产质量管理规范》(2010年修订)中第十章质量控制与质量保证第七节供应商的评估与批准的相关规定。
3.3.13 文件修订的评估 对于2020年版《中国药典》实施后需要修订的文件进行评估。检验方法的标准操作规程可以根据通用检测方法的内容,同时参考《中国药典分析检测技术指南》[4],以及《中国药品检验标准操作规范》的相关内容进行修订。文件的修订还需符合《药品生产质量管理规范》(2010年修订)中第八章文件管理的相关规定。
3.4 开展具体工作 经上述汇总评估,新版药典的整体工作已全部理清,进入具体实施工作阶段,可以启动企业变更控制程序。经过变更评估,评估2020年版《中国药典》变更实施对产品质量的潜在影响。判断变更所需的验证、额外的检验以及稳定性考察应当有科学依据。根据变更评估制定实施计划并明确实施职责,根据计划开展分析方法确认、物料及制剂的再检验、供应商评估、文件及记录的修订、委托检验等相关工作。同时要对相关人员进行2020年版《中国药典》变更内容的培训,使相关人员掌握2020年版《中国药典》的内容,并运用到实际工作中。
4 结果和结论
2020年版《中国药典》与2015年版相比,品种增长5.4%,不但收载品种稳步增加,同时在药品安全性及有效性控制方面的要求不断加强,特别是辅料标准进一步完善和提高,满足了原辅包关联审评审批制度的相关要求。同时2020年版《中国药典》参考人用药品注册技术要求国际协调会(ICH)相关指导原则,修订了分析方法验证、原料药物与制剂稳定性试验等指导原则,不断推进与国际标准协调。
伴随《中国药典》的不断提升,制药企业对于药品的研发、生产、检验等方面也要不断提升自身管理水平。通过比对2015年及2020年版《中国药典》的异同点,对于比对数据分类汇总,并且对不同类型数据进行评估分析,最终确定2020年版《中国药典》实施前制药企业的具体工作。同时参考《药品生产质量管理规范》、《中华人民共和国药品管理法》等相关法规,从而保障2020年版《中国药典》在制药企业的精准实施,从而保障药品的安全、有效、质量可控。同时可以加强药品管理,保证药品质量,保障公众用药安全和合法权益,保护和促进公众健康。
猜你喜欢
承德医学院学报(2022年2期)2022-05-23
医药与保健(2022年2期)2022-04-19
源流(2021年1期)2021-07-28
中学生数理化·高一版(2021年2期)2021-03-19
文萃报·周五版(2020年24期)2020-06-22
中国外汇(2019年21期)2019-05-21
上海人大月刊(2017年9期)2017-10-11
人民周刊(2016年11期)2016-06-30
北方牧业(2016年17期)2016-05-17
新高考·高二数学(2015年11期)2015-12-23
