
溶剂热法制备羧基化超顺磁性Fe3O4纳米颗粒及其磁致变色研究*
2021-02-25 08:21:32许培俊王临江郭新良高尚林
功能材料 2021年1期
许培俊,冯 鑫,王临江,孟 帅,郭新良,高尚林
(1. 长安大学 材料科学与工程学院,西安 710064;2. 云南省电网有限责任公司电力科学研究院,昆明 650217;3. 德国GZD公司,德累斯顿 01187 )
0 引 言
与其他纳米材料一样,Fe3O4纳米颗粒的表面原子数、比表面积和表面能随着粒径的减小逐渐增加,结构单元之间也存在着或强或弱的相互作用,因此Fe3O4纳米颗粒具有表面与界面效应、小尺寸效应和宏观量子隧道效应等。基于以上特性,Fe3O4纳米颗粒呈现出更为优良的电磁、力学、热学、光学等性能。超顺磁性Fe3O4纳米颗粒是指Fe3O4的颗粒尺寸小于临界尺寸时,其各向异性能逐渐减小,Fe3O4纳米颗粒的磁化方向呈现无规律的变化[1]。因此,在外磁场的作用下Fe3O4纳米颗粒的磁化率远大于普通磁性材料,此时Fe3O4纳米颗粒无磁滞现象,剩余磁化强度和矫顽力都趋于零,呈现超顺磁性。与其他磁性纳米颗粒相比,超顺磁性Fe3O4纳米颗粒毒性小具有良好的生物相容性,可通过外磁场作用实现定向移动,因此超顺磁性Fe3O4纳米颗粒在蛋白质分离[2]、肿瘤靶向诊断治疗[3]和磁共振造影成像[4]等医学领域具有广阔的应用前景。同时在外磁场作用下,超顺磁性Fe3O4胶体粒子相互之间会产生磁诱导吸引力;活性基团表面修饰的Fe3O4胶体粒子分散于去离子水中发生电离产生大量的负电荷,表面带有负电荷的Fe3O4胶体粒子相互之间产生静电排斥力。当外磁场强度达到一定范围,致使Fe3O4胶体粒子受到的磁诱导吸引力和静电排斥力相平衡时,超顺磁性Fe3O4胶体粒子能够沿外磁场方向迅速迁移、自组装排列成一维链状光子晶体结构。目前,制备超顺磁性Fe3O4纳米颗粒的方法主要有微乳液法[5]、共沉淀法[6]、溶剂热法[7]等。但是,超顺磁性Fe3O4纳米颗粒在实际应用中暴露出一些问题,如粒径小、比表面积大,且颗粒本身具有磁性容易发生团聚,对环境比较敏感在潮湿环境中易被氧化、稳定性差,在溶剂中分散性差等。为了改善Fe3O4纳米颗粒的应用缺陷,许多学者在制备Fe3O4纳米颗粒的过程中添加适量的有机物[8-9]、无机物[10-11]、高分子聚合物[12-13]等表面活性剂对Fe3O4纳米颗粒进行表面修饰,以此来降低Fe3O4纳米颗粒的表面能,减小颗粒间作用力,制备粒径可控、稳定性好、分散均匀的Fe3O4纳米颗粒。
微乳液法是先将可溶性铁盐和沉淀剂分别制备成水包油型微小液滴,将两种微小液滴在分散相中均匀混合。然后通过机械搅拌使微小液滴之间相互碰撞、破裂、发生反应生成Fe3O4纳米颗粒[14]。采用微乳液法制备Fe3O4纳米颗粒的过程中面临表面活性剂用量大、制备成本高的问题[15]。共沉淀法是在惰性气体如氩气的保护下,向含有铁离子的盐溶液反应体系中加入氢氧化钠、碳酸钠、氨水等共沉淀剂,将混合溶液中的铁离子沉淀出来,再通过热分解等方法生成Fe3O4纳米颗粒[16]。共沉淀法具备制备工艺简便的特点,但反应体系中金属无机盐溶液的种类、溶剂PH、反应温度等因素对Fe3O4纳米颗粒的形貌和粒径分布影响较大,实验重复性差。同时,制备过程中Fe3O4的不均匀沉淀也会使Fe3O4纳米颗粒产生严重的团聚现象。溶剂热法是在密闭的反应容器中采用水或有机溶剂作为反应介质,通过加热提供一个高温高压的反应环境将铁的氢氧化物氧化生成Fe3O4,高温高压的反应环境也可以提高Fe3O4纳米颗粒的纯度及其超顺磁性[17-18]。
柠檬酸、氨基酸等有机小分子中的官能团,通过物理吸附或化学作用键合至Fe3O4纳米颗粒表面对其进行修饰改性。靳艳艳等人[19]将表面油酸充分氧化制备得到粒径大约为12 nm羧基化的Fe3O4纳米粒子,结果表明该Fe3O4纳米粒子在常温下有良好的超顺磁性,同时在去离子水中可长期稳定分散。天然高分子聚合物如壳聚糖、蛋白质和多肽等具有良好的生物相容性和化学稳定性,其分子链上含有丰富的氨基、羟基和羧基等活性基团。马云辉等人[20]首先制备了表面羧基功能化的磁性纳米颗粒,然后通过静电相互作用将壳聚糖自组装在磁性纳米颗粒表面,最后得到粒径大约为10~20 nm并且可在水溶液中形成均匀溶胶的壳聚糖包覆磁性纳米颗粒。
因此,本文以六水合三氯化铁(FeCl3·6H2O)和乙酸钠(CH3COONa)作为原料;HOCH2CH2OH(EG)作为分散介质;C6H5Na3O7·2H2O(Na3Cit)作为表面活性剂,通过溶剂热法制备表面羧基修饰的超顺磁性Fe3O4纳米颗粒。同时,在外磁场的作用下,将特定粒径的超顺磁性Fe3O4纳米颗粒稳定分散于去离子水中制备Fe3O4光子晶体,研究磁场强度对Fe3O4光子晶体结构色的影响。
1 实验部分
1.1 原材料
FeCl3·6H2O(分析纯)、CH3COONa(分析纯)、C6H5Na3O7·2H2O(Na3Cit、分析纯),天津市大茂化学试剂厂;HOCH2CH2OH(EG、分析纯),天津市富宇精细化工有限公司;去离子水(实验室自制)。
1.2 实验过程
1.2.1 Fe3O4纳米颗粒的制备
反应初期,EG作为分散介质为FeCl3·6H2O和CH3COONa的溶解提供液相环境,CH3COONa水解释放出OH-,碱性环境加速FeCl3·6H2O的水解。反应中期,Fe3+和OH-反应生成中间相Fe(OH)3,液相体系中EG将部分Fe3+还原成Fe2+,同时Na3Cit作为静电稳定剂防止磁性二价铁粒子发生团聚。反应后期,Fe(OH)3与Fe(OH)2结合生成Fe3O4,反应方程式如下:
HOCH2CH2OH→CH3CHO+H2O
(1)
CH3COO-+H2O→CH3COOH+OH-
(2)
Fe3++OH-→Fe(OH)3
(3)
CH3CHO+2Fe(OH)3→CH3COCOCH3+2Fe(OH)2+2H2O
(4)
2Fe(OH)3+Fe(OH)2→Fe3O4+4H2O
(5)
称取2.40 g CH3COONa溶于20 mL EG中,恒温40 ℃水浴30 min,使CH3COONa均匀分散于EG中配制A溶液。称取1.08 g FeCl3·6H2O溶于20 mL EG中,同时加入0.6 g Na3Cit配制B溶液,剧烈搅拌待溶液中颗粒物完全溶解,将B溶液缓慢注入A溶液中,40 ℃水浴继续搅拌30 min。将混合均匀的溶液移入容量为100 mL的聚四氟乙烯不锈钢高温高压反应釜中,升温至200 ℃,充分反应。反应结束待反应釜冷却至室温,收集反应釜底部黑色产物,用乙醇和去离子水反复清洗,室温下干燥即制备得到Fe3O4纳米颗粒。
改变反应时间分别为4 、6 、8 、10 h;改变混合溶液中Na3Cit与FeCl3·6H2O的质量比分别为0.28、0.55、0.74,研究反应时间和表面活性剂浓度对Fe3O4纳米颗粒粒径和形貌的影响(表 1)。
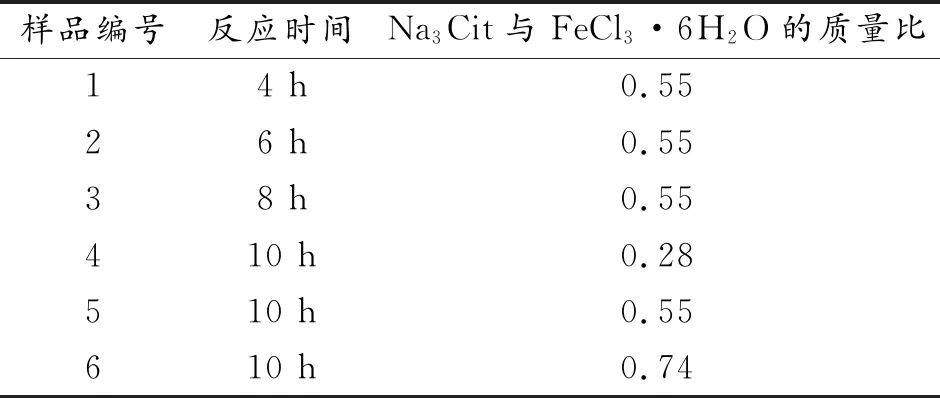
表1 不同反应条件制备Fe3O4纳米颗粒Table 1 Preparation of Fe3O4 nanoparticles under different reaction conditions
1.2.2 Fe3O4光子晶体结构色的制备
特定粒径的超顺磁性Fe3O4胶体粒子离心处理后超声处理1 h分散于去离子水中,配制浓度为10 mg/mL的Fe3O4水分散液。取适量Fe3O4水分散液置于比色皿中,在比色皿侧壁放置一块长方形的永久磁铁使磁场方向垂直于比色皿。通过调节永久磁铁与比色皿的距离定向改变外加磁场强度,研究磁场强度对Fe3O4光子晶体结构色的影响(图 1)。
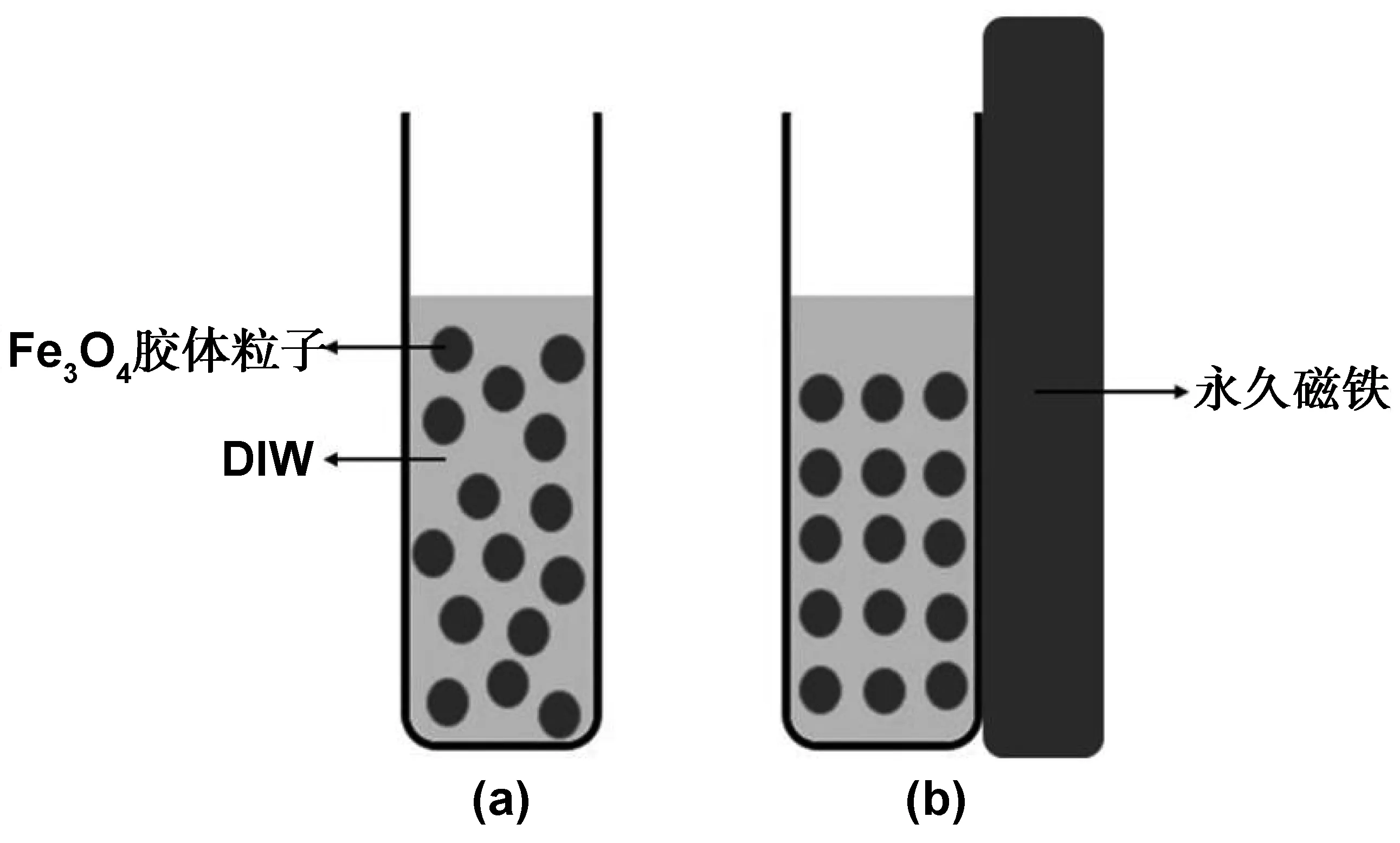
图1 Fe3O4光子晶体磁致变色机理Fig 1 Mechanism of magnetochromism of Fe3O4 photonic crystal
1.3 分析与测试
1.3.1 X射线衍射分析
采用X射线衍射仪(XRD, D8 ADVANCE,德国布鲁克公司)表征Fe3O4纳米颗粒的物相组成。测试时,扫描速度10°/min,扫描范围10~90°。
1.3.2 红外光谱分析
采用傅里叶变换红外光谱仪(FT-IR,TENSOR Ⅱ,德国布鲁克公司)表征Fe3O4纳米颗粒的物相组成,样品制备采用KBr压片法。
1.3.3 热稳定性分析
采用同步热分析仪(SDT650,美国TG公司)表征Fe3O4纳米颗粒的热稳定性。氮气氛围下调节升温速率10 ℃/min,从室温25 ℃升温至900 ℃。
1.3.4 Fe3O4纳米颗粒粒径分布
采用Zeta电位仪(ZEN3600,英国Malvern公司)表征Fe3O4纳米颗粒的粒径分布,样品测试前将浓度为10 mg/mL的Fe3O4水分散液稀释至1 mg/mL。
1.3.5 Fe3O4纳米颗粒微观形貌
采用扫描电子显微镜(SEM,S-4800,日本Hitachi公司)观察分析Fe3O4纳米颗粒的微观形貌,样品表面采用喷金处理。
1.3.6 Fe3O4纳米颗粒磁性能
采用振动样品磁强计(VSM,PPMS-9,美国Quantum Design公司)表征Fe3O4纳米颗粒的磁性能。
2 结果与讨论
2.1 X射线衍射分析
XRD结果表明,通过溶剂热法制备得到反尖晶石结构的Fe3O4纳米颗粒(图 2)。Fe3O4纳米颗粒的XRD谱图中出现了明显的衍射峰,且衍射峰的位置与Fe3O4的PDF标准卡片(85-1436)衍射数据一致,其中2θ=18.285°、30.076°、35.426°、37.057°、43.053°、53.411°、56.935°、62.520°分别对应于Fe3O4的(111)、(220)、(311)、(222)、(400)、(422)、(511)和(440)晶面。这表明采用溶剂法制备的Fe3O4纳米颗粒为反尖晶石结构。根据Scherrer公式计算初始晶粒尺寸:
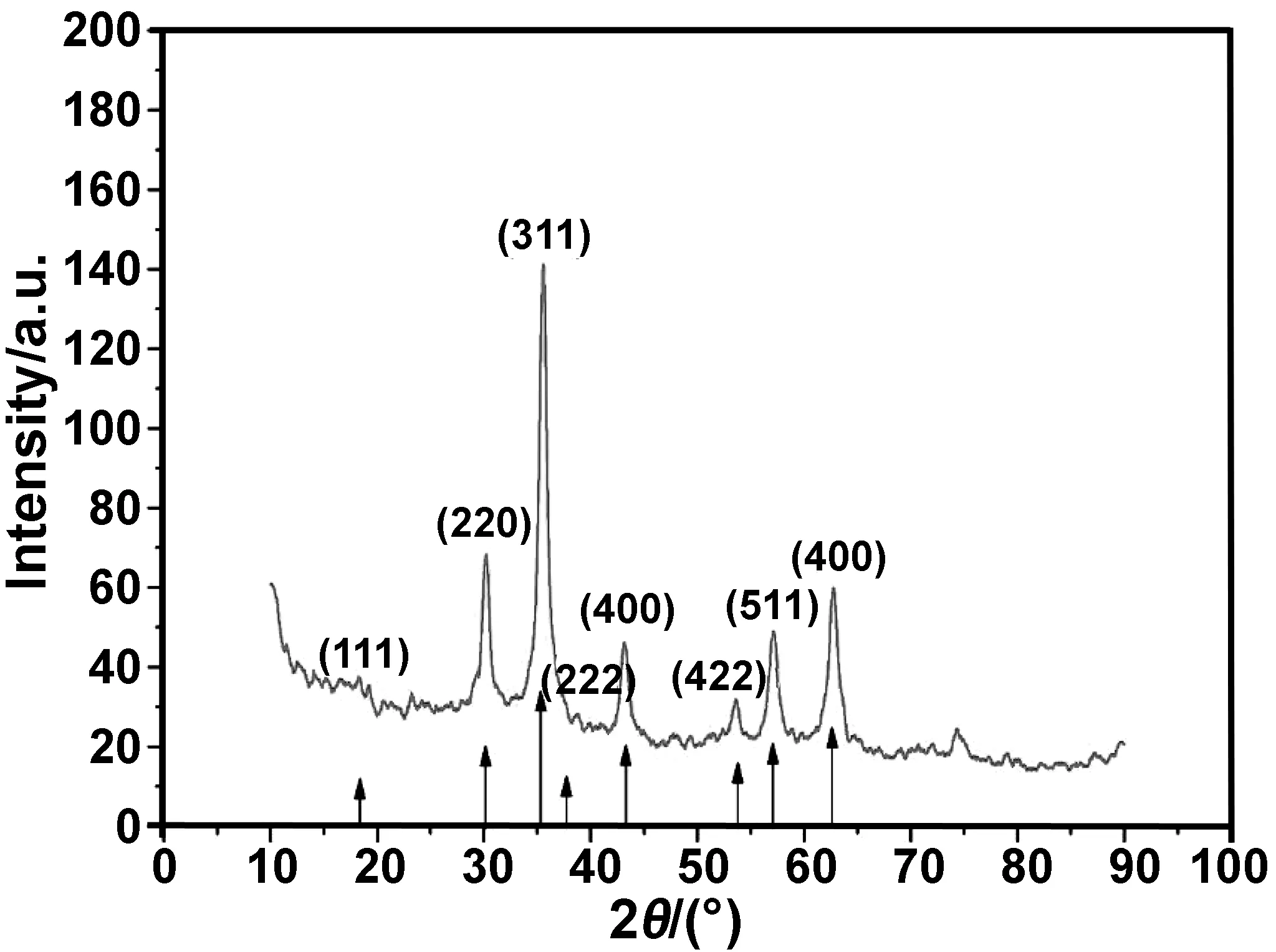
图2 Fe3O4纳米颗粒的XRD谱图Fig 2 XRD spectra of Fe3O4 nanoparticles
(6)
式中,Dhkl为(hkl)晶面的初始晶粒尺寸;k为Scheerer常数,通常k=0.89;λ为入射X射线的波长,测试中Cu靶射线的波长λ=0.154056 nm;β为衍射峰半高宽;θ为布拉格衍射角。以2θ=35.426°为例,计算出(311)晶面对应的初始晶粒尺寸为8.4 nm。
2.2 红外光谱分析
Fe3O4纳米颗粒的红外光谱结果表明,Na3Cit中的官能团键合至Fe3O4纳米颗粒表面,制备得到羧基化的Fe3O4纳米颗粒。Fe3O4纳米颗粒的红外光谱曲线中存在多个吸收振动峰(图 3),580 cm-1处强振动吸收峰对应Fe3O4反尖晶石晶体中Fe—O键的伸缩振动。Fe3O4中Fe—O吸收峰位于570~580 cm-1处,而磁性γ-Fe2O3的特征吸收峰是位于520~610 cm-1处的一个宽峰。由此可分辨Fe3O4和γ-Fe2O3,证实采用溶剂热法制备的黑色产物为纯相Fe3O4纳米颗粒。1 300~1 400 cm-1处是O-H面内形变振动峰,3 400 cm-1左右是Fe3O4吸水峰。1 583 cm-1处对应于CO的伸缩振动峰,1 054 cm-1处对应C—O的伸缩振动。这表明采用溶剂热法制备Fe3O4纳米颗粒的过程中,添加适量的表面活性剂Na3Cit,使得部分羟基、羧基等活性基团键合至Fe3O4纳米颗粒表面(图 4)达到表面修饰的作用,从而提高Fe3O4纳米颗粒的分散性。
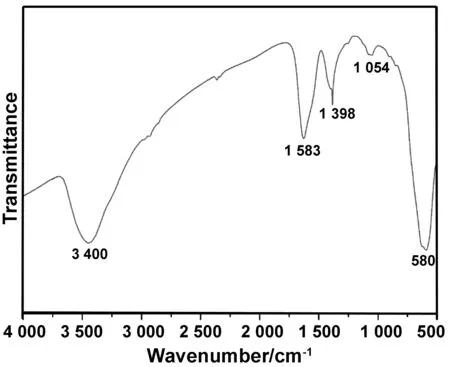
图3 Fe3O4纳米颗粒的红外光谱图Fig 3 Infrared spectrum of Fe3O4 nanoparticles
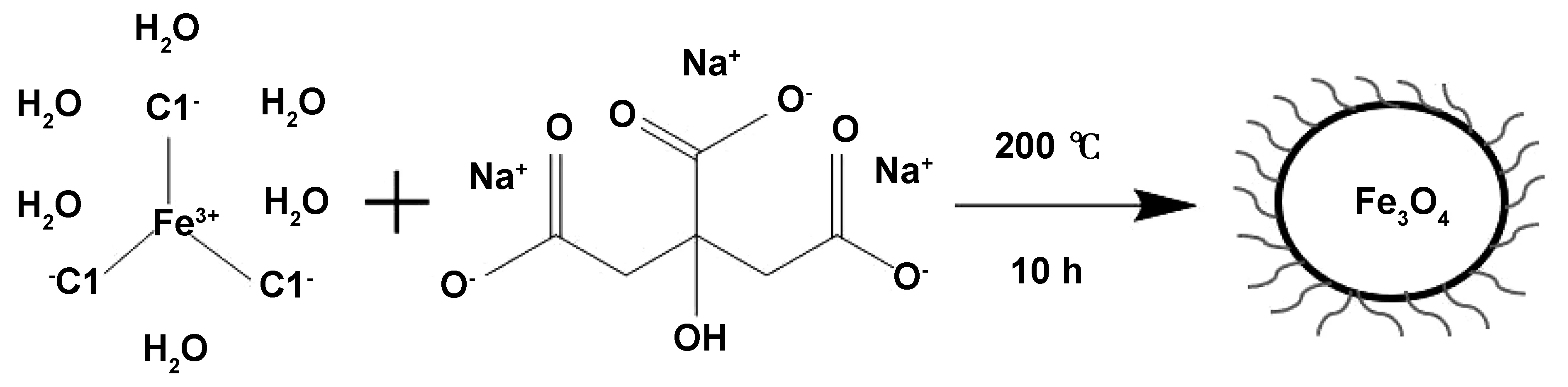
图4 Na3Cit表面修饰Fe3O4纳米颗粒机理Fig 4 Mechanism of surface modification of Fe3O4 nanoparticles by Na3Cit
2.3 热稳定性分析
采用同步热分析仪表征Fe3O4纳米颗粒的热稳定性,其结果表明在逐步升温的过程中Fe3O4纳米颗粒的重量损失达到13%,随着温度继续升高其重量趋于稳定不再急剧下降(图 5)。从室温25℃升温至100℃的过程中,Fe3O4纳米颗粒表面吸附的少量自由水蒸发,致使其重量损失接近3%。100~200 ℃期间出现缓冲区,200 ℃升温至800 ℃的过程中,键合在Fe3O4纳米颗粒表面的有机物受热分解,致使Fe3O4纳米颗粒的重量损失达10%。
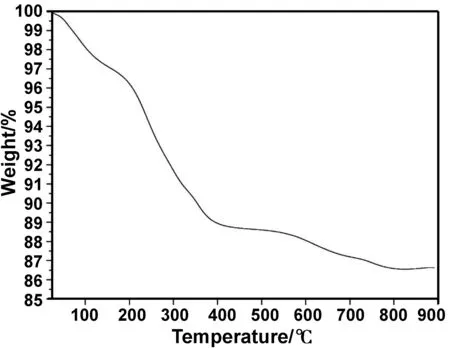
图5 Fe3O4纳米颗粒的TG图Fig 5 TG of Fe3O4 nanoparticles
2.4 反应时间对Fe3O4纳米颗粒粒径和形貌的影响
采用溶剂热法制备Fe3O4纳米颗粒的过程中,Fe3O4纳米颗粒的粒径和形貌与反应时间密切相关,随着反应时间的延长Fe3O4纳米颗粒的粒径逐渐减小(图 6)。反应4 h制备1号样品Fe3O4纳米颗粒的粒径大约为120 nm,颗粒粒径呈正态分布。反应6 h制备2号样品Fe3O4纳米颗粒的粒径大约为100 nm,其SEM结果表明相邻Fe3O4纳米颗粒通过磁性作用相互吸引使得样品出现团聚。反应8 h制备3号样品Fe3O4纳米颗粒的粒径大约为95 nm,样品形貌接近球形。反应10 h制备5号样品Fe3O4纳米颗粒的粒径大约为80 nm,此时Fe3O4纳米颗粒表面比较光滑,呈现完整的球形形貌(图 7)。这是由于,当反应体系中表面活性剂浓度和反应温度一定时,反应时间较短产物反应不完全,导致Fe3O4纳米颗粒粒径不均匀。随着反应时间的延长,液相体系中FeCl3·6H2O和CH3COONa充分水解释放出大量的OH-,促进Fe(OH)3的生成和Fe3+的还原。同时,分散介质EG的黏度较大,降低了Fe3O4纳米颗粒的生长速率。因此,随着反应时间的延长采用溶剂热法制备的Fe3O4纳米颗粒其粒径大大减小。
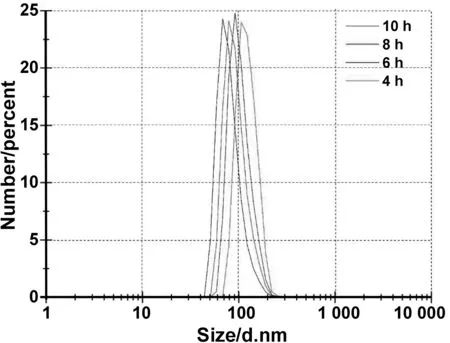
图6 反应时间分别为4、6、8 、10 h制备的Fe3O4纳米颗粒的粒径分布图Fig 6 The particle size distribution of Fe3O4 nanoparticles prepared with reaction time of 4, 6 , 8 and 10 h

图7 反应时间分别为4h (a)、6h (b)、8h (c)、10h (d)制备的Fe3O4纳米颗粒的SEM图Fig 7 SEM of Fe3O4 nanoparticles prepared with reaction time of 4 , 6 , 8 and 10 h
2.5 Na3Cit浓度对Fe3O4纳米颗粒粒径和形貌的影响
采用溶剂热法制备Fe3O4纳米颗粒的过程中,随着反应体系中表面活性剂Na3Cit的浓度逐渐增大,Fe3O4纳米颗粒粒径呈逐渐减小的趋势(图 8)。当反应体系中Na3Cit与FeCl3·6H2O的质量比为0.28,升温至200 ℃反应10 h,经洗涤、干燥后Fe3O4纳米颗粒粒径大约为110 nm(图 9(a))。这是由于液相环境中表面活性剂浓度太小时,Fe3+和Fe2+并没有被表面活性剂形成的水核包覆,而是以离子的形式自由存在,因此形成的Fe3O4纳米颗粒粒径较大。反应体系中Na3Cit与FeCl3·6H2O的质量比为0.55,升温至200 ℃反应10 h,经洗涤、干燥后Fe3O4纳米颗粒粒径大约为80 nm(图 9(b))。采用溶剂热法制备Fe3O4纳米颗粒的过程中,随着反应体系中Na3Cit浓度增大,Na3Cit释放出的羟基和羧基逐渐增多。羧基与Fe3+具有很强的配位作用,致使Fe3O4纳米颗粒表面的负电荷增多,静电斥力和表面张力的共同作用抑制Fe3O4晶体进一步长大,致使Fe3O4纳米颗粒粒径逐渐减小。反应体系中Na3Cit与FeCl3·6H2O的质量比为0.74,升温至200℃反应10 h,制备的Fe3O4纳米颗粒粒径分布较广,其对应的SEM结果表明此时粒径大小分布不均匀(图 9(c))。这是由于反应体系中表面活性剂浓度太大时,过量的表面活性剂造成部分Fe3O4纳米颗粒相互黏结,从而引起粒径分布较广,难以控制。
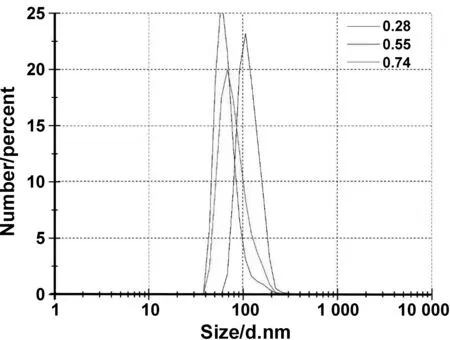
图8 Na3Cit与FeCl3·6H2O的质量比分别为0.28、0.55、0.74制备的Fe3O4纳米颗粒的粒径分布图Fig 8 The particle size distribution of Fe3O4 nanoparticles prepared by the mass ratio of Na3Cit to FeCl3·6H2O is 0.28, 0.55 and 0.74

图9 Na3Cit与FeCl3·6H2O的质量比分别为0.28 (a)、0.55 (b)、0.74 (c)制备的Fe3O4纳米颗粒的SEM图Fig 9 SEM of Fe3O4 nanoparticles prepared by the mass ratio of Na3Cit to FeCl3·6H2O of 0.28, 0.55 and 0.74
2.6 Fe3O4纳米颗粒的磁性能
反应体系中Na3Cit与FeCl3·6H2O的质量比为0.55,升温至200 ℃反应10 h,制备的Fe3O4纳米颗粒的磁滞回线结果表明(图 10):室温下该Fe3O4纳米颗粒的饱和磁化强度为60.149 A·m2/kg,其剩余磁化强度和矫顽力均趋近于零,样品具有良好的超顺磁性。
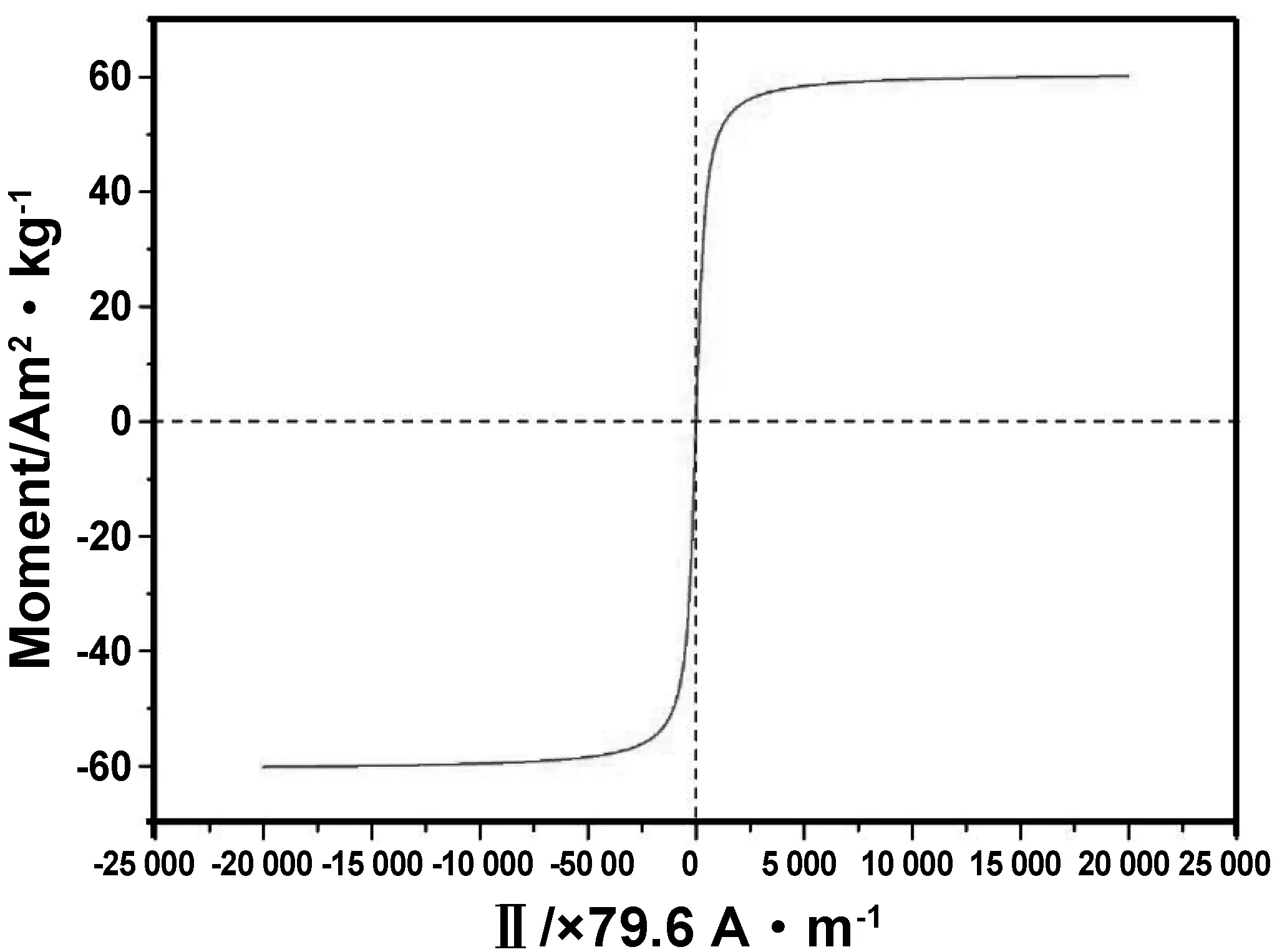
图10 Fe3O4纳米颗粒的磁滞回线Fig 10 Hysteresis loops of Fe3O4 nanoparticles
基于以上讨论,确定采用溶剂热法制备Fe3O4纳米颗粒的最佳实验条件为:混合溶液中Na3Cit与FeCl3·6H2O的质量比为0.55,升温至200℃反应10 h。通过该反应条件可制备得到粒径为80 nm的超顺磁性Fe3O4纳米颗粒。
2.7 磁场强度对Fe3O4光子晶体结构色的影响
将3 mL浓度为10 mg/mL的Fe3O4水分散液置于比色皿中,没有外磁场作用时,溶剂中的Fe3O4胶体粒子一直处于无规则的布朗运动中(图 1(a))。当一定强度的外磁场作用时,Fe3O4胶体粒子沿磁场方向自组装成周期性排列(图 1(b)),光在这种周期性有序结构中的传播遵循布拉格衍射定律。因此,当特定频率的入射光落在光子带隙中时,会得到明亮的反射光谱,从而实现Fe3O4光子晶体的磁致变色。通过调整比色皿与永久磁铁之间的距离改变磁场强度,研究外加磁场强度对Fe3O4光子晶体结构色的影响。结果如图 11所示,比色皿与永久磁铁间距离分别为0、2、3、4、5 mm时,对应的Fe3O4光子晶体结构色分别为蓝色、蓝绿色、绿色、青绿色、黄绿色。当比色皿距离永久磁铁较远时(≥6 mm)时,Fe3O4光子晶体不再呈现明亮的结构色。根据布拉格衍射方程:

图11 从左到右比色皿与永久磁铁的距离分别为0、2、3、4、5、6 mmFig 11 The distance between the colorimetric plate and the permanent magnet from left to right is 0, 2, 3, 4, 5, 6 mm
mλ=2dsinθ
(7)
其中m为衍射级数;λ为反射光的波长;d为晶面间距;θ为布拉格衍射角。
装有适量Fe3O4水分散液的比色皿与永久磁铁间距离逐渐增大时,溶剂中相邻Fe3O4胶体粒子间的磁诱导吸引力逐渐减小。为了使Fe3O4胶体粒子受到的磁诱导吸引力和静电排斥力相平衡,相邻Fe3O4胶体粒子间距离d逐渐增大,以此来减小粒子间静电排斥力。因此,对于特定粒径的Fe3O4胶体粒子,光子晶体结构色的可调性与磁场强度有关,随着磁场强度B逐渐减小,Fe3O4光子晶体反射峰位置发生红移,逐渐向长波长方向移动。但是,通过不同反应时间4 、6、8 h制备的粒径分别为120、100、95 nm的Fe3O4纳米颗粒,将其超声分散于去离子水中配制相同浓度的Fe3O4水分散液,采用相同的方法诱导其自组装并没有产生明亮的光子晶体结构色,这可能是因为Fe3O4胶体粒子颗粒尺寸分布不均匀、形貌不规整等原因影响其紧密堆积自组装。
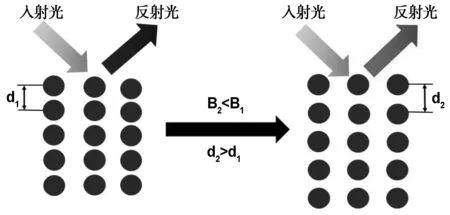
图12 磁场强度对Fe3O4光子晶体结构色的影响Fig 12 Effect of magnetic field intensity on structure color of Fe3O4 photonic crystal
3 结 论
本文采用溶剂热法制备了分散性良好的超顺磁性Fe3O4纳米颗粒,研究表明反应时间和表面活性剂浓度对Fe3O4纳米颗粒的粒径均存在显著影响。同时通过添加表面活性剂C6H5Na3O7·2H2O,在Fe3O4表面键合部分活性基团,改善了Fe3O4纳米颗粒的分散性和稳定性。通过外磁场诱导致使超顺磁性Fe3O4胶体粒子定向排列自组装形成Fe3O4光子晶体,研究磁场强度对Fe3O4光子晶体结构色的影响。结果表明,超顺磁性Fe3O4胶体粒子不仅在水分散液中可以通过磁诱导形成一维光子晶体,同时可以通过调节外磁场强度定向调控胶体粒子间距,从而调控光子带隙构筑磁响应性光子晶体。
猜你喜欢
光子学报(2022年11期)2022-11-26 03:43:44
中国民间疗法(2021年6期)2021-06-09 06:18:58
科技创新导报(2019年11期)2019-07-13 09:40:56
医学研究杂志(2015年3期)2015-06-10 06:41:52
湖南师范大学自然科学学报(2015年1期)2015-02-27 14:50:05
应用化工(2014年12期)2014-08-16 13:10:46
机械制造与自动化(2014年1期)2014-03-01 04:21:57
原子与分子物理学报(2014年4期)2014-02-28 22:18:47
无机化学学报(2014年6期)2014-02-28 17:32:06
无机化学学报(2014年6期)2014-02-28 17:31:57
