
WO3 增强RuO2/ZrO2 表面酸性及其NH3 选择性催化氧化性能研究
2021-02-24 06:15:38李粉吉张艳琨杨春晓张可欣夏福婷张秋林庞鹏飞王慧敏
燃料化学学报 2021年2期
李粉吉 ,张艳琨 ,杨春晓 ,张可欣 ,夏福婷,2,* ,张秋林 ,庞鹏飞 ,王慧敏
(1.云南民族大学 云南省高校民族地区资源清洁转化重点实验室,云南 昆明 650500;2.昆明理工大学 省部共建复杂有色金属资源清洁利用国家重点实验室,云南 昆明 650093;3.昆明理工大学 环境科学与工程学院,云南 昆明 650093)
在现代工业活动中,选择性催化还原(SCR)技 术是从固定源中消除NOx使用较为广泛的一种方法。在NH3-SCR 系统中,需要加入过量的氨气来满足NOx还原的目标,然而,这些未反应的NH3会直接逃逸到大气中,对生态环境和人体健康造成潜在的危害[1,2]。因此,去除NH3-SCR 反应系统中多余的NH3是很有必要的。目前,用于消除NH3的技术主要有吸附法、吸收法、催化分解法和选择性催化氧化法。其中,选择性催化氧化(NH3-SCO)法是将NH3与O2在有催化剂存在的情况下转化为对人体健康和环境无危害的氮气和水,从而达到去除NH3的目的[3],其优点是操作简单,效率高,被认为是去除NH3的比较有前景的方法之一[4-8],催化剂的选择是这项技术的核心。
目前,用于NH3-SCO 的催化剂主要有分子筛催化剂,负载型贵金属催化剂和过渡金属催化剂。分子筛催化剂主要包括Cu-ZSM-5、Fe-ZSM-5和Fe-Beta,大部分分子筛催化剂表现出较好的氮气选择性,但反应条件较为苛刻,一般需要较高的温度才能实现氨的完全转化[9-11]。负载型的贵金属催化剂主要有Ag/Al2O3[12]、CuO/RuO2[13]、Pt/CuO/Al2O3[14]和Ir[15]等,这些催化剂大部分在低于180 ℃时即可以获得较高NH3转化率,但N2选择性有待进一步提高。常见的过渡金属氧化物主要包括Fe2O3、CuO、NiO、V2O5、Co3O4和MnO2等,研究者们已将这些催化剂作为NH3-SCO 技术中的潜在催化剂[16-21]。其中,研究较多的为铜基催化剂,因为其具有高N2选择性和较好的稳定性。然而,在这些催化剂上NH3能够完全转化的温度大多在300 ℃以上[22-25],催化活性一般没有贵金属负载型催化剂优异。Liang 等[26]采用浸渍法制备了Cu/γ-Al2O3催化剂,并将其应用于NH3-SCO 中,研究发现氮气选择性高达95%,但是大约在400 ℃才能实现氨的完全转化。Chmielarz 等[27]通过共沉淀法制备了不同Cu 含量的Mg-Cu-Fe 催化剂,根据活性测试结果发现,Cu 加入后催化剂的活性有所提升,但是NH3大约在400 ℃才能实现完全转化。Sazonova 等[28]研究表明,Cu/TiO2催化剂的N2选择较好,但在300-400 ℃才能实现NH3的完全转化。虽然大量文献报道过渡金属氧化物催化剂活性好,氮气选择性好,但一般来说,贵金属催化剂的低温活性还是要优于过渡金属催化剂。
为了兼顾氮气选择性的同时提高催化活性,不少研究者在过渡金属催化剂上进行了改性形成复合金属催化剂。一般认为,复合金属氧化物之间由于存在结构或电子调变等相互作用,活性比相应的单一氧化物要高[24]。Zhang 等[29]将过渡金属与贵金属结合起来,采用溶胶凝胶法制备了RuO2-Fe2O3催化剂,将不同含量的RuO2与Fe2O3相结合,之后用WO3改性催化剂,增强了催化剂的表面酸性位点,做到优势互补,催化剂的活性和氮气选择性有所提升。Yang 等[30]结合Cu 基催化剂氮气选择性高和Ag 基催化剂的低温活性的特性,制备了Cu-Ag/A12O3催化剂,用于NH3-SCO 技术中,研究结果表明,氨气在低于320 ℃时就可以完全转化,并且氮气选择性可以达到95%。
研究发现,三氧化钨(WO3)作为重要的催化剂助剂具有较好的热稳定性,不仅可充当酸性组分,而且还能提高催化剂的稳定性[31]。Wang 等[32]发现,将WO3引入到RuO2-Fe2O3催化剂中增强了氨的吸附能力,并提高了高温N2的选择性。Ma等[33]发现,WO3-ZrO2载体具有较高的稳定性和较强的表面酸度。但在NH3-SCO 中并没有人对RuO2负载到载体WO3-ZrO2上进行研究。为了研究固体酸对NH3-SCO 中提高氮气选择性能的影响,采用不同含量的WO3来对RuO2/ZrO2催化剂进行改性,采用浸渍法制备了一系列的RuO2/WO3-ZrO2催化剂,并将其应用于NH3-SCO 中,通过不同的表征手段来分析RuO2/WO3-ZrO2催化剂上NH3氧化的反应机理及其氮气选择性提升的关键所在。
1 实验部分
1.1 催化剂的制备
1.1.1 ZrO2 和WO3-ZrO2 载体的制备
为了探究钨酸在NH3-SCO 中对氮气选择性提升性能的影响,采用WO3对催化剂进行处理。采用共沉淀法制备了ZrO2和不同WO3含量的WO3-ZrO2载体,WO3的含量分别为5%、10%、15%和20%。制备流程如下:首先将一定量的Zr(NO3)2·5H2O 分别溶于去离子水中,然后分别加入所需含量的偏钨酸铵不断搅拌直至完全溶解。之后逐滴滴加氨水至溶液pH 值达到10 左右,稳定30 min左右。将得到的悬浊液在室温下静置2 h,然后在80 ℃水浴中老化4 h,静置过夜后抽滤,用去离子水洗涤数次,105 ℃烘箱中干燥24 h,之后将样品在马弗炉中空气气氛下550 ℃焙烧3 h,升温速率为2 ℃/min。样品被标记为ZrO2和xWO3-ZrO2(x表示WO3的质量分数,x= 5%、10%、15%、20%)。
1.1.2 RuO2/ZrO2 和RuO2/WO3-ZrO2 催化剂的制备
RuO2/ZrO2和RuO2/xWO3-ZrO2催 化 剂 采 用 过量浸渍法制备,活性前驱体盐为RuCl3·5H2O,RuO2的负载量低至0.5%。将配置好的活性前驱体盐溶于一定量的去离子水中,称取一定量的载体倒入溶液中在室温下不断搅拌2 h,静置2 h 后,在70 ℃水浴中搅拌6 h 后,置于105 ℃烘箱中干燥12 h,之后在马弗炉中焙烧3 h,焙烧温度为500 ℃,升温速率为1 ℃/min。为了描述方便,所制备的催化剂分别被标记为Ru/Zr 或Ru/xW-Zr (x代表WO3的质量分数x= 5%、10%、15%、20%)。
1.2 催化剂表征
X 射线衍射技术已经成为最基本、最重要的一种结构测试手段,主要用来对物质进行物相分析,测定物质的结晶度。样品的XRD 分析是在BrukerD8 ADVANCE A25X 衍射仪记录上进行的,射线源为CuKα(λ=0.15406 nm),工作电压为40 kV,工作电流为40 mA,步长为0.02°,扫描速率为6 (°)/min,10°-90°扫描。
催化剂的平均孔径使用Barrett-Joiner-Halenda(BJH)模型计算得到,催化剂的比表面积通过Brunauer-Emmett-Teller(BET)方法获得。样品的N2吸附-脱附等温线是在Quantasorb Junior 吸附仪(TriStar II 3020)上进行测试。在测试之前,将样品在氮气氛围下于300 ℃吹扫2 h。
H2-程序升温还原采用自制的包含石英管反应器的吸附装置测试。采用GC9750 型色谱仪,热导检测器(TCD)进行信号采集。首先称取50 mg 催化剂置于石英管中,将石英管放于反应炉内,为了除去催化剂表面的杂质,首先将催化剂在氮气氛围下以8 ℃/min 的升温速率升温至400 ℃进行吹扫,在400 ℃阶段保持40 min,N2的流量为30 mL/min。然后让炉子温度降至50 ℃以下,在30 mL/min H2/N2(H2= 5%)的气流下升温至700 ℃,升温速率为8 ℃/min。
有艺术悟性的刘曼文,用绘画语言和人们交流,她很低姿态,有时很羞怯,但也很自信,甚至很倔强,人们之所以乐意接受她和她的艺术,因为她和它们真诚。
X 射线光电子能谱实验(XPS)是在Thermo Scientific K-Alpha 型电子能谱仪上进行XPS 测试的,激发源为AlKα,样品进行抽真空处理,分析室真空度优于10-8Pa,结合能校正:以C 1s= 284.80 eV 结合能为能量标准。
催化剂的Raman 表征采用英国Renishaw 公司生产的inVia 型拉曼光谱仪测试。激光器的波长为532 nm,激光功率 ≥ 50 mW,测量波数为100-1200 cm-1。
1.3 催化剂活性评价
NH3-SCO 活性测试是在固定床石英管(长600 mm,内径6 mm)反应器中进行,该反应器以稳定流动模式进行。每个样品催化剂用量为0.4 mL,活性测试温度窗口在150-350 ℃,每隔25 ℃监测一个点,达到指定温度后稳定30 min 时再采集气体进行分析。入口处的混合气体包括5%的O2、0.08%的NH3,N2作为平衡气体。进口处气体的总流量为400 mL/min,气空时速为60000 h-1。形成的NO和NO2气体使用烟气分析仪(ECOM·J2KN)测定。N2O 的含量是通过带有电子捕获检测器的气相色谱(GC-9790)进行检测。NH3的含量由氨气分析仪(GXH-1050E,北京)测定。NH3转化率和氮气选择性分别通过以下公式计算得到:
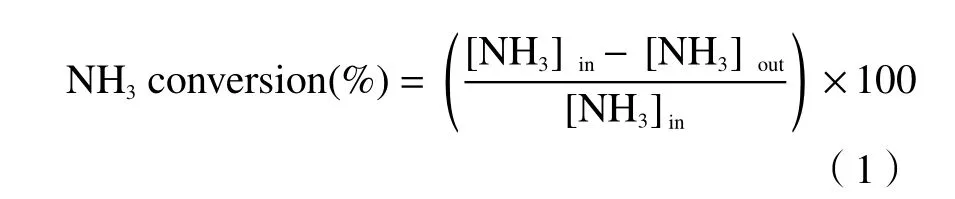
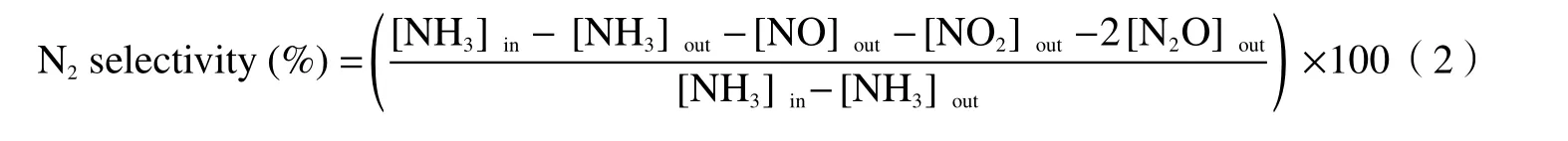
2 结果与讨论
2.1 不同含量WO3 掺杂对Ru/Zr 催化剂上NH3-SCO 催化性能的影响
笔者对所有的催化剂进行了活性测试,相应的结果见图1。由图1(a)可知,所有的催化剂都表现出良好的低温活性,NH3完全转化的温度都不超过250 ℃。其中,Ru/Zr 催化剂在225 ℃能够实现NH3的完全转化,但是由图1(b)可知,Ru/Zr催化剂上副产物的产率相对较高。在Ru/Zr 催化剂中加入WO3之后,所有Ru/W-Zr 催化剂的高温N2选择性均有所提高。当WO3的含量为5%和10%时,在提高催化剂N2选择性的同时不会降低活性。随着WO3含量增加至15%和20%时,催化剂的活性会稍微有所降低,大约在250 ℃时实现100%的氨气转化率,但是从氮气选择性的结果来看,催化剂中WO3的含量增加至15%和20%时,相比于Ru/10%W-Zr 而言其氮气选择性也没有提升太多。基于以上结果可知,适量WO3的引入不仅可以保持Ru/Zr 催化剂本身优异的催化活性,还可以显著提高催化剂高温氮气选择性。所以结合活性测试和氮气选择性可以知道WO3最佳的含量为10%。

图1 Ru/Zr 和不同含量WO3 掺杂的Ru/Zr 催化剂的NH3-SCO 性能测试Figure 1 NH3-SCO performance test of Ru/Zr and Ru/Zr catalysts doped with different contents of WO3(a): NH3 conversion; (b): N2 selectivity
2.2 XRD 结果讨论
为了研究WO3改性对Ru/Zr 催化剂结晶度和晶型的影响,对所有样品进行了XRD 表征分析,结果见图2。在不含钨的Ru/Zr 的衍射图中观察到四方晶型ZrO2(t-ZrO2)和单斜晶ZrO2(m-ZrO2)的特征衍射峰[23]。其中,位于24.19°、28.13°、31.27°、34.21°、40.89°、55.41°、45.21°、65.82°处的衍射 峰归属于四方晶型ZrO2(m-ZrO2)的特征峰,在35.20°、50.12°、59.93°处的特征峰归属于单斜晶ZrO2(t-ZrO2)的特征峰。当用WO3改性后,物种的晶型发生了明显的改变。当WO3的含量为5%时,在30.09°处出现了t-ZrO2的衍射峰,而m-ZrO2的衍射峰的峰强度也逐渐减弱;当WO3的含量增加到10%的时候,只在28.13°处观察到一个很弱的m-ZrO2物种的衍射峰;当WO3的含量增加到15%时,m-ZrO2物种的衍射峰几乎探测不到,仅观察到t-ZrO2的特征峰,这可能是因为m-ZrO2物种太少无法被探测到。这和之前的报道[34,35]相符合,加入过WO3可以使m-ZrO2物种向热力学更稳定的t-ZrO2物种转变,因为单斜晶t-ZrO2在酸性中心的形成中起重要作用,所以加入WO3有利于提高催化剂的稳定性[36,37]。在所有样品中均未发现WOx的特征峰,表明形成了WOx-ZrO2固溶体[34,35]。据报道,只有当温度高于900 ℃时才会出现WO3的晶体结构(23.2°、23.7°、24.3°)[38,39]。这可能是在Ru/xWO3-ZrO2催化剂的XRD 谱图中未观察到WO3特征峰的主要原因。根据XRD 可以推测出,t-ZrO2物种的形成增加了催化剂的稳定性,在催化剂表面有利于氨的吸附,不利于氨与氧快速反应,相应的氮氧化物会减少,氮气选择性就会提升。
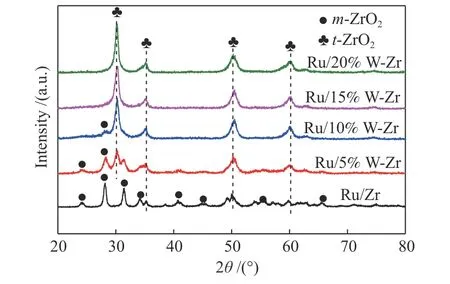
图2 Ru/Zr 和不同含量WO3 掺杂的Ru/Zr 催化剂的XRD 谱图Figure 2 XRD spectra of the Ru/Zr and WO3 doped Ru/Zr catalysts
2.3 催化剂的N2 吸附-脱附分析
催化剂的N2吸附-脱附曲线和孔径分布见图3。由图3(a)可知,Ru/Zr 催化剂和WO3改性的Ru/Zr催化剂均具有典型的“IV”型吸附等温线,表明催化剂具有介孔(2-50 nm)结构[40]。Ru/Zr 催化剂的滞回环出现在p/p0= 0.7-1.0 附近,属于H3 类型。当催化剂用WO3改性后,Ru/xW-Zr 催化剂的滞回环出现在p/p0= 0.4-1.0 附近,由H3 类型变为H1 类型。和Ru/Zr 催化剂相比,Ru/xW-Zr 催化剂中滞回环的闭合点存在于较低的p/p0(0.4)处,表明WO3的加入能使催化剂形成更多孔径稍小的介孔[41]。由孔径分布图(图3(b))可知,Ru/Zr 催化剂的解吸孔径分布主要集中在15 nm 附近,而Ru/xW-Zr 催化剂的孔径主要分布在2-8 nm,表明WO3的加入使催化剂的孔径分布变得更小。
表1 为Ru/Zr 催化剂和不同含量WO3改性Ru/Zr 催化剂的BET 比表面积、孔径和孔容分布。Ru/Zr 催化剂的BET 比表面积,孔体积和平均孔径分别为40.7 m2/g、0.21 cm3/g 和14.5 nm,Ru/5%W-Zr催化剂的BET 比表面积为68.1 m2/g,孔体积和平均孔径分别为0.19 cm3/g 和7.2 nm,和Ru/Zr 催化剂相比,Ru/5%W-Zr 催化剂的BET 比表面积有所增加,孔体积和平均孔径均有所减小。随着WO3含量的逐渐增加,催化剂的BET 比表面积逐渐增大,孔体积和平均孔径均逐渐减小,表明钨和锆物种之间的相互作用改善了催化剂的孔径,进而导致催化剂有较大的BET 比表面积。较高的BET 比表面积有助于NH3或NOx的吸附,不利于氨与氧发生快速反应,这也是氮气选择性提升的一个主要原因[42]。此外,与Ru/Zr 催化剂相比,钨改性的催化剂孔体积和平均孔径都有所减小,表明钨的引入有利于催化剂孔径分布向小孔方向发展,进而增大其比表面积。
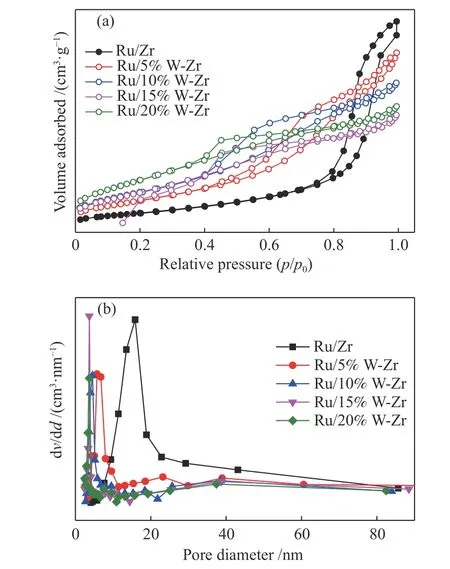
图3 Ru/Zr 和不同含量WO3 掺杂的Ru/Zr 催化剂的N2吸附-脱附曲线和孔径分布Figure 3 N2 adsorption-desorption curve and pore size distribution of Ru/Zr and WO3 doped Ru/Zr catalysts
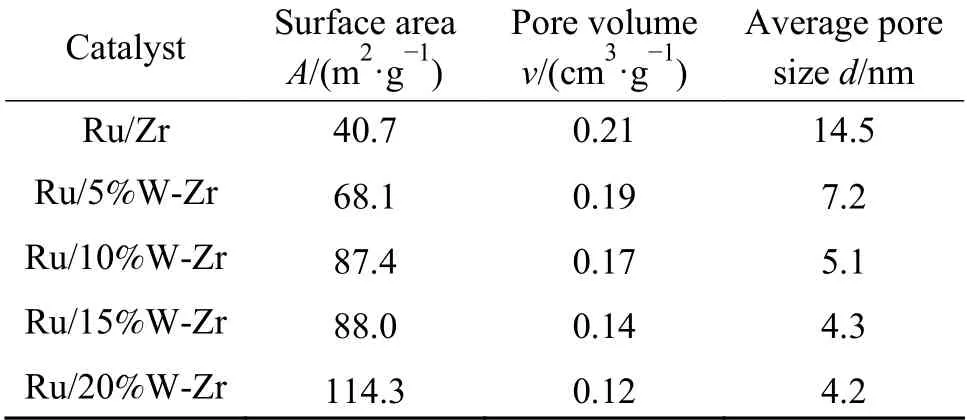
表1 Ru/Zr 和不同含量WO3 掺杂的Ru/Zr 催化剂的孔结构参数Table 1 Pore structure parameters of Ru/Zr and WO3 doped Ru/Zr catalysts
2.4 拉曼(Raman)光谱
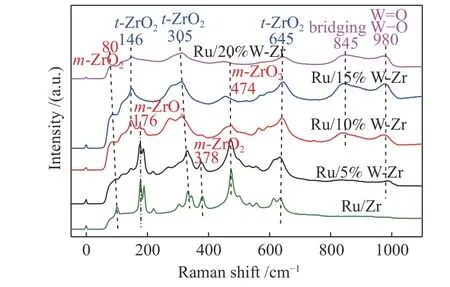
图4 Ru/Zr 和不同含量WO3 掺杂的Ru/Zr 催化剂的拉曼光谱图Figure 4 Raman spectra of Ru/Zr and WO3 doped Ru/Zr catalysts
2.5 H2-TPR 表征
众所周知,催化剂的氧化还原性在NH3-SCO反应中扮演着重要的角色,为了研究WO3的加入对催化剂氧化还原性能的影响,对所有催化剂进行了H2-TPR 测试,结果如图5 所示。对于WO3-ZrO2载体,在H2-TPR 谱图中并未检测到任何WO3物种的还原峰,表明有效的W-O 和Zr-O键在低于700 ℃的温度条件下无法被H2还原。在Ru/Zr 或Ru/xW-Zr 催化剂的H2-TPR 谱图中均有还原峰被检测到,这些低温还原峰均属于不同状态的RuO2的还原,说明RuO2物种比较容易被还原[46],这也说明贵金属催化剂RuO2的活性较好。对于Ru/Zr 催化剂,在105 ℃左右出现了还原峰,而Ru/5%W-Zr、Ru/10%W-Zr、Ru/15%W-Zr 和Ru/20%W-Zr 催化剂的还原峰分别出现在温度为136、139、162 和166 ℃处。显然,WO3的加入使催化剂的还原峰峰位置发生了偏移,随着WO3含量的增加还原峰的位置逐步向高温方向移动,表明WO3物种与RuO2物种之间存在较强的相互作用,增加了催化剂的稳定性,其氧化还原能力降低。出色的氧化还原能力可能会导致NH3过度氧化和NOx的形成,从而使N2选择性降低[47]。然而,WO3的加入使催化剂变得相对稳定,其氧化还原能力降低,催化剂上合适的氧化还原性能可以抑制NH3过渡氧化,减少NOx的形成,从而提高催化剂的高温N2选择性。
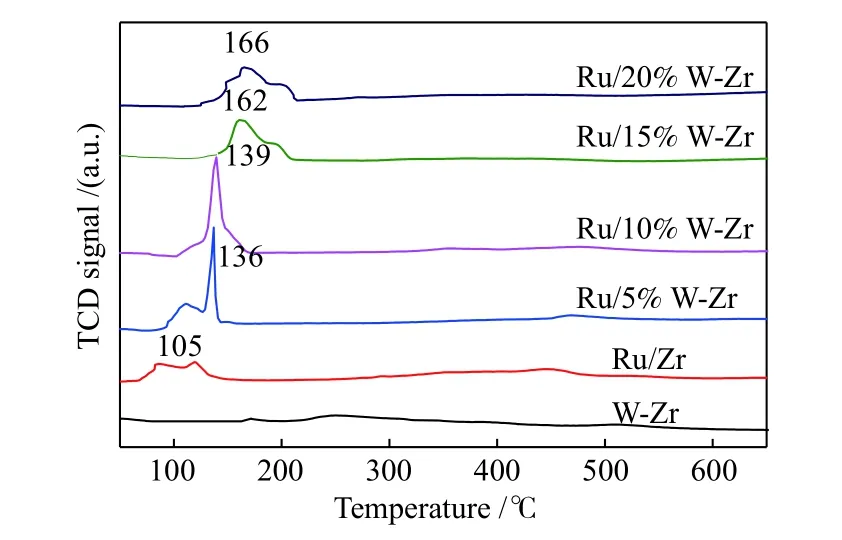
图5 载体WO3-ZrO2、Ru/Zr 和不同含量WO3 掺杂的Ru/Zr 催化剂的H2-TPR 谱图Figure 5 H2-TPR spectra of the WO3-ZrO2 support, Ru/Zr and WO3 doped Ru/Zr catalysts
2.6 XPS 表征
XPS 技术用于探究所有金属的化学态以及表面组成信息,为了获得Ru/Zr 和Ru/15%W-Zr 催化剂的表面组成和化学价态信息,Ru/Zr 和Ru/15%W-Zr催化剂的XPS 检测结果见图6 所示。由图6(a)可知,两个催化剂的Ru 3p光谱在结合能463.2 和461.9 eV 处出现了两个主要的信号峰,证实了Ru4+和Ru0物种的存在[48]。对比两个样品能明显看出Ru/Zr 样品种表面Ru4+含量比Ru/W-Zr 样品多,一般来说Ru4+在NH3-SCO 反应中起到十分重要的作用,因此,Ru/Zr 表现出较为优异的氧化还原性能(还原峰温度最低)和催化活性;此外,从图6(b)中O 1s的XPS 光谱中也能看出,Ru/Zr样品中表面氧尤其是活性氧要比Ru/W-Zr 多,这也突出其优异的氧化还原性能(还原峰温度最低)和催化活性,但是氧化性过强又会导致NH3发生过氧化产生更多副产物,因此,W 的作用就是减弱这种氧化作用,提高催化剂表面酸性,在保证活性的同时提高N2选择性[49]。
2.7 催化剂的原位DRIFT 光谱
2.7.1 NH3 的吸附
众所周知,在NH3-SCO 中催化剂的表面酸性起很大的作用,为了进一步研究催化剂表面酸性位点的类型,测试了Ru/Zr 和Ru/15%W-Zr 催化剂在不同温度下NH3吸附的原位DRIFTS,结果见图7。由图7(a)可知,对于Ru/Zr 催化剂,当温度升至50 ℃时,通入氨30 min 后有多个信号分别在3369、3232、1823、1604、1556、1475、1437、1315、1179 和1085 cm-1处被检测到,其中,3369 和3232 cm-1处的信号可归属于NH3中N-H 键的伸缩振动模式。在1604和1179 cm-1处出现的谱带归属于Lewis(L)酸性位点上的配位NH3物种[31,50]。1823 cm-1处的峰属于-HNO 物种,随着反应温度的升高,峰信号逐渐减弱,到350 ℃完全消失,这可能是因为吸附的NH3物种首先在催化剂表面通过脱氢作用生成酰胺(-NH2)和酰亚胺(-NH)(反应(3)-(4)),然后酰亚胺(-NH)物种被氧化形成-HNO(反应(5)-(6))[31]。当温度升高时,亚硝基(-HNO)物种又被氧原子氧化为NO(反应(7))[31,32]。位于1475 和1437 cm-1处三个谱带归属于吸附在Brønsted(B)酸性位点上的物种[51]。1556 和1085 cm-1处的特征峰归属于酰胺(-NH2)物种[52]。从图中可以明显看出,吸附在Lewis(L)酸 性 位 点(1604 和1179 cm-1)上 的NH3物种随着温度的升高而迅速消失。而吸附在Brønsted( B) 酸 性 位 点 上 的物 种 的 信 号(1475 和1437 cm-1)随温度的升高而逐渐降低,这表明吸附在L 酸性位点上的NH3要比吸附在B 酸性位点上的NH3更容易反应。随着温度升高,在1556 和1085 cm-1处的峰强度随着温度的升高稍微有所增强。表明在反应过程中随着温度的升高NH3物种发生了脱氢作用,生成-NH2物种(反应(3))。

Ru/15%W-Zr 催化剂最终的DRIFTS 列在图7(b)中。在温度为50 ℃的条件下,在Ru/15%W-Zr 催化剂上吸附8×10-4的NH3后,四个信号比较强的峰在1675、1601、1439 和1234 cm-1处出现,三个信号比较弱的峰在波数分别为3349、3261 和1557 cm-1处出现。其中,3349 和3261 cm-1处的特征谱带归属于NH3中N-H 的伸缩振动模式。在1675 cm-1处有一个比较平缓的特征峰,归属于化学吸附在Brønsted(B)酸性位点上的对称伸缩振动[32]。1601 处有一个比较尖锐的特征峰,可归属为Lewis(L)酸性位点上的配位氨分子,1557 cm-1处的特征峰属于酰胺(-NH2)物种,表明WO3改性后催化剂表面仅仅发生了脱氢作用。在1439 和1234 cm-1处的特征峰分别归属于Brønsted 酸和Lewis(L)酸。
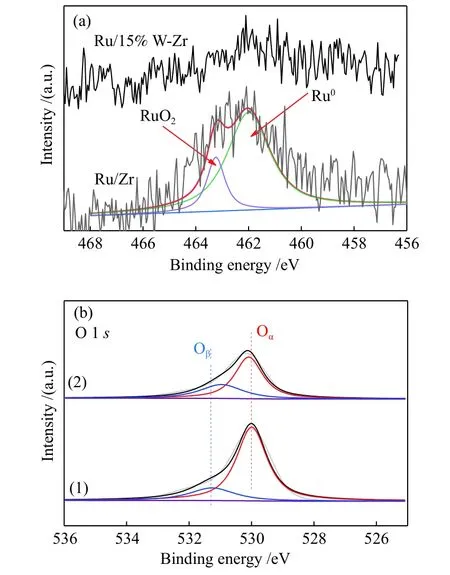
图6 催化剂Ru/Zr 和Ru/15%W-Zr 催化剂的XPS 谱图Figure 6 XPS spectra of the Ru/Zr and Ru/15%W-Zr catalysts

图7 (a)催化剂Ru/Zr 和(b)Ru/15%W-Zr 催化剂的表面酸性信息图Figure 7 Surface acidity of (a) Ru/Zr catalyst and(b) Ru/15%W-Zr catalyst
与Ru/Zr 催化剂相比,用WO3处理的Ru/15%W-Zr 催化剂上原位红外谱图中的Lewis 和Brønsted酸性位点谱带信号明显增强,中间物种明显减少,表明酸改性的催化剂可以促进氨物种的吸附。由此可知,催化剂表面适当的酸度可以抑制氨过氧化,从而减少副产物的形成,使N2选择性得到明显提升。
为了研究催化剂表面的化学反应过程,分别对Ru/Zr 和Ru/15%W-Zr 在200 ℃温度下进行了in-situDRIFTS 分析。样品在200 ℃温度下用8×10-4的NH3预处理30 min,然后通入氮气吹扫,再将5%的O2通入原位池,在不同的时间段采集光谱图,相应的结果见图8。对于Ru/Zr 催化剂而言,相应的DRIFT 光谱见图8(a)。用NH3处理30 min 后,在3354 和3246 cm-1分别有两个特征峰,归属于NH3中N-H 的伸缩振动模式,1821 cm-1处的特征峰属于-HNO 物种,1602 和1181 cm-1处的峰信号属于Lewis(L)酸性位点上的配位氨分子。1471 cm-1处的峰信号归属于吸附在Brønsted(B)酸性位点上的对称伸缩振动。显然,当通入5%的O21 min时,3354、3246、1602和1181 cm-1处的峰信号迅速消失,1821 cm-1处的特征峰偏移至1842 cm-1处,一个新信号立即出现在1583 cm-1处。毫无疑问,在1842 cm-1处的峰信号归属于-HNO 物种,1583 cm-1处的特征峰可归属于硝酸盐类[53,54]。两个新形成的物种的谱带强度随NH3通入时间的延长逐渐增强。这些现象表明,随着气态氧的通入,催化剂表面吸附氨物种会与大量氧反应,从而导致硝酸盐物种和亚硝酰基物种的形成。
对于Ru/15%W-Zr 催化剂,在200 ℃下预吸附的NH3与O2反应的DRIFT 光谱分别见图8(b)。首先,从吸附氨到通氧的整个反应过程中,没有新的特征峰出现,1672 cm-1处的特征峰在通入氧后迅速消失。3350、3268、1604 和1238 cm-1处的峰信号属于吸附在L 酸性位点上的配位NH3物种。1672 和1430 cm-1处的特征峰属于B 酸性位点上的物种,只有少量的氨活化中间物种-NH2(1540 cm-1)被检测到。从图中可以清楚地看出,这些谱带随着反应时间的延长几乎没有变化,这是由于Ru/15%W-Zr 催化剂表面的酸性较强,吸附在L 酸性位点和B 酸性位点上的NH3物种较稳定。公认的是,酸性较强可以抑制NH3在较低温度下发生活化。
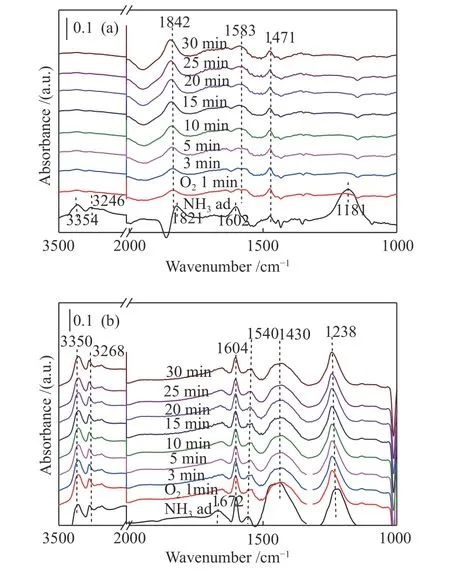
图8 200 ℃下Ru/Zr(A)和Ru/15%W-Zr(B)催化剂表面反应信息图Figure 8 Surface reaction of the Ru/Zr(A) and Ru/15%W-Zr(B) catalysts at 200 ℃
根据上述结果,与Ru/Zr 相比,WO3的掺杂有利于氨物种的吸附,不利于氨与氧气发生快速反应,可以有效地抑制氮氧化物的形成。这也是WO3的引入导致氮气选择性提高的主要原因。根据以上结果可以推断出发生在WO3改性的催化剂上的氨氧化反应过程遵循内部选择性催化还原(i-SCR)机理,以下是在催化剂表面可能发生的反应途径:
2.8 催化剂的稳定性测试
长期稳定性测试对于评估一个催化剂的应用价值是十分重要的。因此,本研究选取Ru/Zr和Ru/15%W-Zr 催化剂对其进行了长期催化稳定性测试。测试条件为:将催化剂在混合气氛(8×10-4NH3,5% O2和N2平衡气)下250 ℃进行50 h 的活性测试。测试结果见图9,Ru/Zr 和Ru/15%W-Zr催化剂的NH3转化率在长期的活性测试期间保持稳定。总的来说,在250 ℃,50 h 的测试时间内实现了100%的NH3转化率。因此,Ru/Zr 和Ru/15%WZr 表现出优异的催化稳定性,结合氮气选择性可知Ru/15%W-Zr 是理想的NH3-SCO 催化剂。
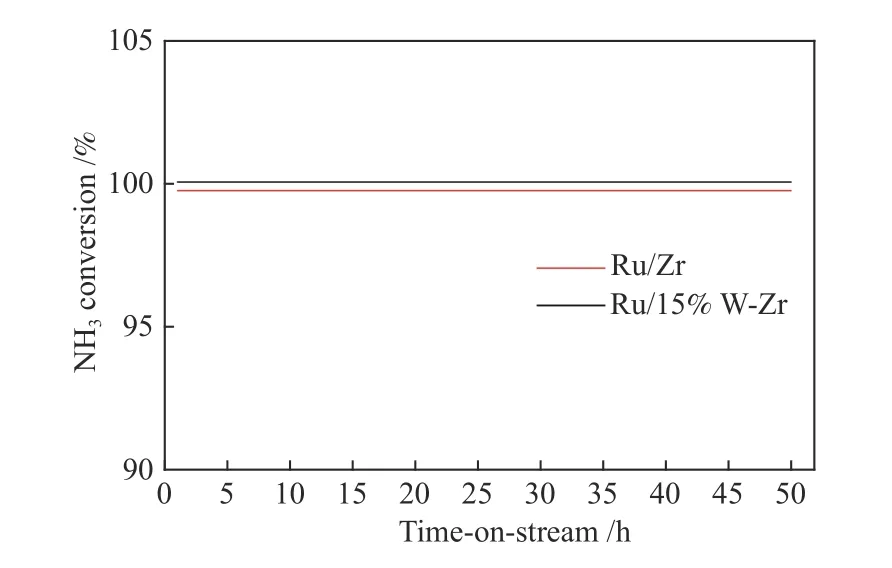
图9 Ru/Zr 和Ru/15%W-Zr 催化剂催化性能的长期稳定性测试Figure 9 Dependence of catalytic performance on reaction time of the Ru/Zr and Ru/15%W-Zr catalysts
3 结 论
本研究主要研究了WO3的加入在NH3-SCO反应中对氮气选择性提升性能的影响。虽然纯的Ru/Zr 催化剂在较低的温度条件下就能表现出优越的催化性能,但其氮气选择性并不是很理想,适量WO3的加入能够在其活性几乎保持不变的情况下对氮气选择性的提升起到促进作用,WO3的含量太高(20%)会使催化剂的活性下降,而氮气选择性在WO3的含量为10%的基础上没有太大变化,所以WO3的最佳含量为10%。此外,通过一系列的表征分析可以发现,WO3和ZrO2以及RuO2之间存在较强的相互作用,WO3的加入能够使ZrO2的晶型发生改变,提高催化剂的稳定性,使催化剂的氧化还原能力降低,表面酸性位点增强。这些都有利于氨物种在催化剂表面的吸附,抑制氨与氧发生快速反应,这是氮气选择性提升的主要原因。
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:28
第一财经(2019年8期)2019-08-26 17:53:46
福建基础教育研究(2019年8期)2019-05-28 08:39:51
中学生数理化·八年级物理人教版(2017年6期)2017-11-09 06:00:43
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:52
当代化工研究(2016年5期)2016-03-20 16:21:32
哈尔滨医药(2015年2期)2015-12-01 03:57:13
学习月刊(2015年14期)2015-07-09 03:37:48
物理化学学报(2015年5期)2015-02-28 17:34:47
天然气勘探与开发(2015年1期)2015-02-28 17:00:44
