
X-蜡生成过程中自由基产生的分子动力学仿真
2021-02-23 07:15沈殷和齐波林元棣张毅涛
电力工程技术 2021年1期
沈殷和, 齐波, 林元棣, 张毅涛
(1. 新能源电力系统国家重点实验室(华北电力大学),北京 102206;2. 华北电力大学高电压与电磁兼容北京重点实验室,北京 102206;3. 国网江苏省电力有限公司电力科学研究院,江苏 南京 211103)
0 引言
油纸绝缘结构设备在解体时常有黄色蜡状沉积物存在。这种沉积物没有固定的化学表达式,被称作X-蜡[1]。X-蜡通常出现在变压器、电流互感器和套管等油纸绝缘结构中[2—3],并伴有大量故障特征气体。马卫平等人在高压电流互感器解体过程中,发现绝缘层多处存在X-蜡[4—5];甘德刚等人对500 kV换流变压器网侧故障套管进行解体时,在导电杆上发现X-蜡[6]。Orlando Girlanda等人通过实验测量,发现X-蜡会使得50 Hz下介损增大,导致整体损耗增大,局部温度上升[7]。但目前还未有针对X-蜡产生机理的详细报道。
近年来ReaxFF力场[8]被广泛应用于描述复杂化学反应过程。文献[9]采用ReaxFF反应力场构建山茶籽绝缘油模型,模拟高温下热解和产气的规律。文献[10—12]利用ReaxFF力场对绝缘纸和纤维素的高温裂解过程进行研究。文献[13]基于ReaxFF力场对聚酰亚胺高温下绝缘失效的微观机理进行了模拟分析。研究证明,ReaxFF-MD方法可以动态地描述反应物成键断键过程,揭示产物的生成路径,为X-蜡产生的动态模拟提供良好基础。
文中基于分子动力学采用ReaxFF力场模拟绝缘油在温度473 K,393 K,373 K下自由基的产生过程,并分析蜡的形成方式和组成成分,为更好地理解X-蜡产生机理奠定了基础。
1 绝缘油分子动力学仿真
1.1 仿真对象
油纸绝缘结构中常用的绝缘油型号为25号,故文中仿真采用的绝缘油模型构建借鉴了刘枫林等人利用质谱仪测得的新疆克拉玛25号环烷基矿物油成分组成,链烃和环烷烃占环烷烃矿物油总质量的88.6%。具体分布如下:链烃质量分数为11.6%;环烷烃质量分数为77.0%,其中一环烷烃15.5%,二环烷烃28.5%,三环烷烃23.3%,四环烷烃9.7%[14]。
环烷基油作为混合物,把油中所有成分的分子结构和在油中的含量表示出来是没有必要且不现实的。环烷烃分子在环烷基油中占主要部分,模拟过程中只要能够充分体现出环烷基的物化特性即可。根据所测得的组分分布,可以将整个油模型等效为5种烃类分子组成[15]。即链烃C12H26,一环烷烃C14H28,二环烷烃C13H24,三环烷烃C16H28和四环烷烃C16H26,其形态如图1所示。

图1 环烷基油模型的5种单体分子Fig.1 Five monomer molecules of naphthenic oil model
1.2 仿真环境及过程
在分子动力学软件中构建绝缘油模型,并模拟烃类分子在不同温度下的分解情况,步骤如下:
(1) 根据环烷基油的5种主要组成部分,搭建5种单体分子的三维模型,然后利用Forcite模块分别对各个分子进行几何结构优化,优化力场采用适用于有机小分子和高分子的COMPASS力场。
(2) 利用 Amorphous Cell 模块建立不定形液态系统,液体密度设为0.2 g/cm3。液体系统共包含 30 个分子[16],依据矿物绝缘油中各成分的质量分数计算出分子个数。计算步骤如下:
(1)
(2)
式中:m(CxHy)为各组分的分子量;ω(CxHy)为各组分所占质量分数。
由此计算出链烃、一环烷烃、二环烷烃、三环烷烃和四环烷烃的个数分别为5,5,10,7,3。构建完成后得到初步模型,如图2所示。可以看到,在初步模型中,分子在盒子中分布不均匀,导致低密度区域的形成,要解决这个问题,必须对该液体系统进行优化,执行短暂的能量最小化过程,并施加一个分子动力学弛豫过程来平衡盒子[17—19]。
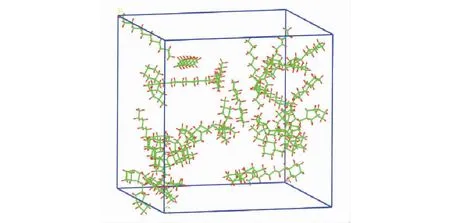
图2 构建初期盒子构型Fig.2 Early configuration of the cell
(3) 动力学弛豫过程主要分为3步。① 对系统进行退火。在周期性变化的温度下运行分子动力学进行弛豫处理, 在NVT(分子数、体积和温度不变)正则系统下,将退火循环数设置为5,将初始温度设置为300 K,中循环温度设置为500 K。② 在NPT(分子数、压强和温度不变)正则系统下,对系统进行加压和解压处理,加压时设置压强为0.1 GPa,解压时设置压强为0.1 MPa。③ 利用Forcite模块中的Dynamics功能,使系统在NVT正则系统下达到300 K下的平衡状态,平衡时系统密度达到了0.82 g/cm3,平衡状态下油模型如图3所示。

图3 平衡状态下油模型Fig.3 The oil model in equilibrium
1.3 步长、温度及其他参数的选择
在分子模拟中,积分步长的选择十分重要,太长的时间步长会造成分子间的激烈碰撞,导致体系数据溢出;太短的时间步长会降低模拟过程搜索相空间的能力;一般选取的时间步长为体系各个自由度中最短运动周期的1/10[20]。根据红外光谱中的波数可以计算出化学键的频率:
(3)
式中:k为化学键的力常数,与键能和键长有关,具体见表1;μ为双原子折合质量,μ=m1m2/(m1+m2),m1,m2为原子的分子量。

表1 不同种类碳氢键对应的力常数Table 1 Force constants of hydrocarbon bonds
从而计算得到绝缘油模型中最小振动周期:
(4)
(5)
T=1/f=1.08×10-14s
(6)
式中:c为光速。
取振动周期的1/10作为积分步长,所以选择积分步长为1 fs。若积分步长过小,2次相邻计算间分子的运动变化很小,会导致在设置的模拟时间内得不到想要的结果。若积分步长过大,就会使仿真过程中温度急剧上升,体系数据溢出导致仿真终止。
目前研究显示,X-蜡在较低温度下就能产生。Orlando Girlanda等人解体故障套管时在绝缘纸上发现了X-蜡,经过绝缘纸的DP测量发现绝缘纸并没有发生分解[7],而变压器中常用的B级绝缘纸在130 ℃的条件下,纤维素中葡萄糖键就会断开发生分解[21],可见X-蜡在130 ℃以下就能够产生。温度过高也不利于X-蜡的形成,文献[5]认为X-蜡由碳原子数为12~24的烷烃组成,而文献[22]在热解理论中提到,温度超过200 ℃时,C10以上的烷烃会全部受热分解,所以温度过高并不利于大分子的X-蜡形成,文中将仿真的上限温度设为200 ℃。文献[1]指出110 ℃是矿物油中产生X-蜡的阈值温度,低于阈值温度无法产生X-蜡。综合考虑,文中选择仿真温度为473 K,393 K和373 K。
采用Gulp模块的Dynamics功能进行模拟仿真,观察液态体系在不同温度下的分解动态变化过程。力场选择ReaxFF6.0,系统设置为NVT正则系统,仿真时间为100 ps。仿真结束后会生成xtd格式的轨迹文件以及仿真过程中温度、能量和压力随时间变化的曲线,可以通过播放轨迹文件观看各分子的运动情况和化学键的断裂情况。
2 仿真结果分析
ReaxFF力场是根据各粒子间的距离来确定相互间的作用力强弱,可以很好地表示化学键成键和断键情况。文中根据分子间的距离判断化学键是否断裂。当化学键两原子间距离扩大为原来的5倍时,则认为化学键已经断裂。
经过多次仿真并通过轨迹文件捕捉到473 K下第一个自由基的产生,如图4所示,发生分解的分子为四环烷烃C16H26。

图4 473 K下第一个自由基的产生Fig.4 First free radical generation at 473 K
从图中可以看到,四环烷烃五元环上的碳氢键发生断裂。观察温度变化趋势图和轨迹文件,可知在前15 ps,分子主要处于振动过程而没有发生化学键的断裂;在15 ps左右,化学键开始发生断裂。
393 K下的结果与473 K下相似,但是分解速度相比于473 K情况下慢了很多,化学键的断裂发生在90 ps左右。通过轨迹文件捕捉到393 K下第一个自由基的产生,如图5所示,发生分解的分子同样为四环烷烃C16H26。
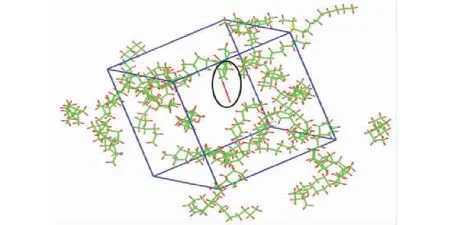
图5 393 K下第一个自由基的产生Fig.5 First free radical generation at 393 K
373 K下的仿真结果对比393 K和473 K下的结果有些不同。在仿真时间100 ps内并没有发生分解,延长仿真时间发现在130 ps左右发生化学键断裂。捕捉到的率先发生分解的分子为三环烷烃C16H28。第一个自由基的产生情况如图6所示。
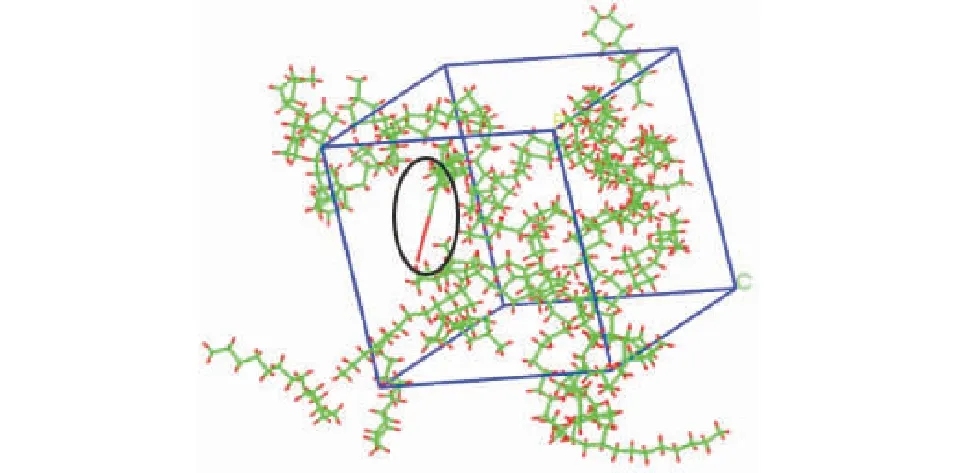
图6 373 K下第一个自由基的产生Fig.6 First free radical generation at 373 K
对比不同温度下化学键断裂的情况,发现环烷烃的碳氢键更容易发生断裂,当碳氢键发生断裂后就会形成自由基,即一个较大分子量的烃类自由基和一个氢自由基。由于自由基的化学性质很活泼,自由基之间很容易相互作用形成新的产物,2个氢自由基相互作用会形成氢气;氢自由基和烃类自由基作用会重新形成环烷烃,再次进入反应当中;2个烃类自由基相互作用形成不溶于油的X-蜡,所以X-蜡分子量也会大于组成绝缘油的烃分子。
为验证该观点,采集500 kV主变套管导电杆上的X-蜡试样,采用vario EL cube元素分析仪通过燃烧法进行元素分析[23],结果如表2所示。

表2 元素分析结果Table 2 Chemical element analysis
通过元素分析法获得的蜡状物的C、H、O 元素质量比,计算C、H、O元素原子数比为 20∶35∶0.4,即最简式为C20H35O0.4。考虑到仪器的测量误差在 1%~3%范围内, 因而忽略氧元素,获得蜡状物的元素 C、H 比例为20∶35。通过元素分析,推断该套管蜡状物主要成分是 C、H,其最简式是为C20H35。可以看到这种X-蜡的分子量大于绝缘油中的母体烃。
而且由于不同环烷烃之间碳氢键键能的差距不明显,在外界因素作用下,会有多种烷烃发生分解并形成不同种类自由基,自由基的相互组合是完全随机的,存在多种链终止反应[24],会生成多种大分子产物,导致X-蜡以混合物形式存在。
仿真结果表明,473 K,393 K,373 K温度下,第一个自由基产生时间分别为15 ps,90 ps,130 ps。可以看到,在仿真温度范围内,温度越高,第一个自由基产生的时间越短。
3 结论
文中基于分子动力学建立了含有30个分子的矿物绝缘油仿真模型,在ReaxFF力场中模拟473 K,393 K,373 K下油模型中X-蜡产生过程的自由基生成情况,结论如下:
(1) X-蜡生成第一步为化学键的均裂形成2个自由基,在仿真温度范围内,自由基产生速度与温度呈正相关,通过仿真发现这个过程主要发生在环烷烃分子上。环烷烃分子上碳氢键易发生断裂形成2个自由基,2个大分子量的自由基重组后就会形成X-蜡。
(2) 仿真过程中步长应该设置为体系各个自由度中最短运动周期的1/10,积分步长选择过小,2次相邻计算间分子的运动变化很小,会导致在设置的模拟时间内得不到想要的结果。积分步长选择过大,就会使仿真过程中温度急剧上升,体系数据溢出导致仿真终止。
猜你喜欢
石油炼制与化工(2022年6期)2022-06-21
中国音乐学(2022年1期)2022-05-05
煤炭转化(2020年2期)2020-04-24
物理学报(2018年10期)2018-06-14
润滑油(2016年4期)2016-11-04
考试周刊(2016年60期)2016-08-23
考试周刊(2016年48期)2016-06-29
中学生数理化·高二版(2016年6期)2016-05-14
润滑油(2015年3期)2015-08-08
化学教学(2014年1期)2014-02-20
