
儿童肾透明细胞瘤的临床病理特征
2021-02-23 08:52阿智祥谢余澄李霁伟武红芳
检验医学与临床 2021年4期
周 军,段 玲,高 燕,阿智祥,谢余澄,李霁伟,武红芳
1.昆明市儿童医院病理科,云南昆明 650028;2.云南省第二人民医院检验科,云南昆明 650034
肾透明细胞瘤(CCSK)是一种十分罕见的儿童恶性肾肿瘤,约占小儿肾肿瘤的2%~5%,好发于6个月至5岁的婴幼儿,平均发病年龄在3岁左右[1],1970年由Kidd首次命名,将其从肾母细胞瘤(Wilms瘤)分离出来成为一个独立实体肿瘤。CCSK是一种具有高度侵袭性,可广泛转移的恶性肿瘤,尤其好发骨转移,其预后较差、病死率较高,目前国内外尚无明确的诊疗指南。临床上该肿瘤发病年龄、病变部位及影像学检查等无特异性,术前常诊断为肾母细胞瘤,术后病理诊断时尤其应注意与单一分化的肾母细胞瘤和先天性中胚叶肾瘤鉴别,目前CCSK并没有特异性的诊断标志物,为此,本研究通过回顾分析昆明市儿童医院2010年1月至2019年12月收治的11例CCSK患儿的临床及病理资料,探讨其临床及病理特征、免疫组化表型、鉴别诊断,现总结报道如下。
1 资料与方法
1.1一般资料 收集昆明市儿童医院2010年1月至2019年12月收治的11例CCSK患儿的临床及病理资料。
1.2方法 将CCSK的临床及病理表现,包括发病年龄、性别、术前影像学资料、肿瘤大小、转移情况、病理形态及免疫组化结果等进行对比分析,探讨CCSK的临床及病理表现特点。免疫组化染色采用EnVision两步法,一抗、二抗及显色系统试剂均购于基因科技(上海)股份有限公司。
1.3统计学处理 采用SPSS21.0统计软件进行数据处理及统计学分析。
2 结 果
2.1临床特征 本组CCSK患儿主要为婴幼儿,患儿发病年龄为1岁至7岁9个月,中位年龄24个月,男女比例为1.0∶1.2,主要临床症状表现为腹部包块、腹部疼痛及肉眼血尿。本研究中11例患儿有5例发生转移或侵犯,其中1例发生颅内转移,1例发生盆底转移,3例发生肾周或下腔静脉的转移及侵犯。11例患儿的临床资料见表1。CCSK的影像学检查无明显特异性,增强CT见图1,11例患儿术前均被诊断为Wilms瘤。
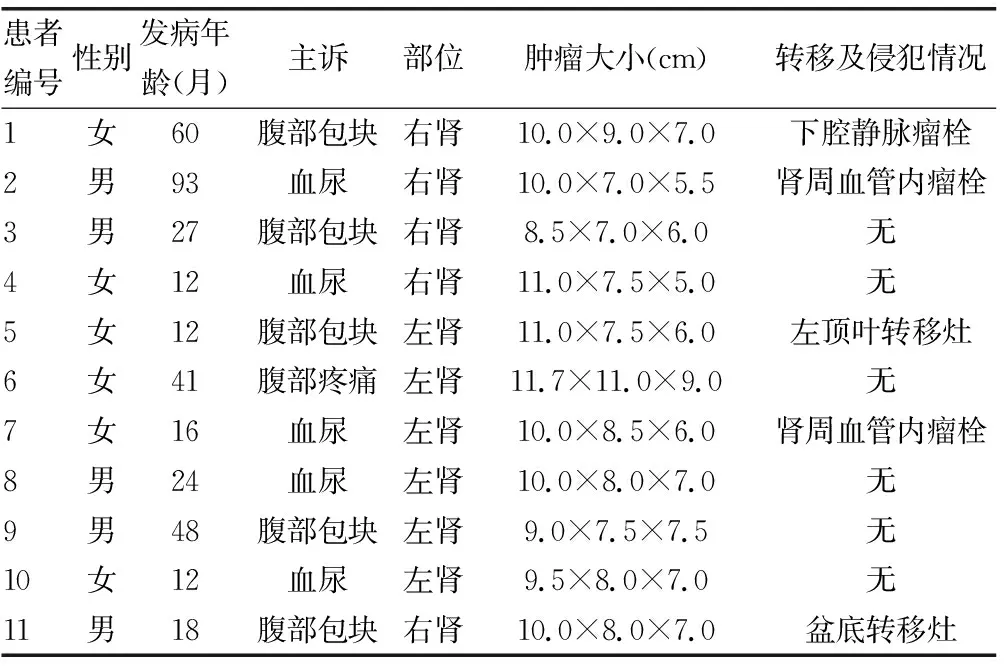
表1 11例CCSK患儿的临床资料
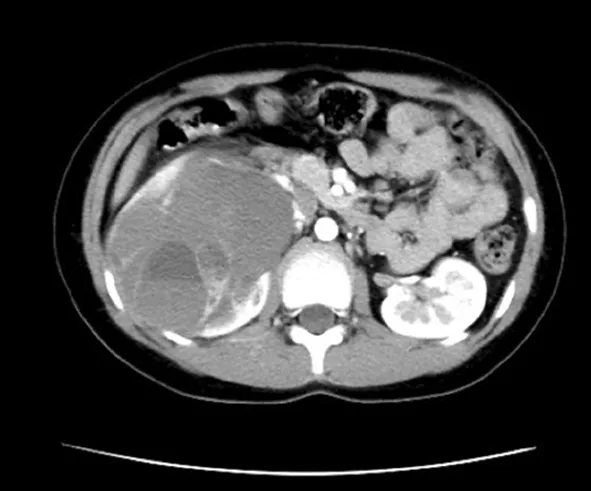
注:肿瘤位于右肾上极,右肾及右侧肾上腺区可见一较大不规则形混杂密度灶,增强扫描呈不均匀强化,可见斑片状液化坏死区,边界尚清。图1 CCSK的影像学检查
2.2病理形态特点
2.2.1大体检查 肿瘤体积一般较大,肿瘤平均最大径为(10.06±0.91)cm,没有明显包膜,边界尚清,切面实性,灰白色或棕褐色,鱼肉状或黏液透明样,可见囊性变,有的区域为编织状,质地柔韧,常伴有坏死区域。
2.2.2镜下检查 经典的CCSK细胞呈巢状或者条索状排列,周围间质有树枝状纤维血管将其分隔,肿瘤细胞呈上皮样或梭形,被细胞外黏液样物质松散地分开,胞质较空淡,几乎不着色,呈透明细胞状,见图2A。细胞核呈棒状、卵圆形或略不规则,染色质细腻,核仁不明显,核膜薄,少数核有明显核沟。间质较薄,其间见规则分枝状的“鸡爪样”毛细血管,见图2B,或较厚的纤维母细胞条索围绕着毛细血管,部分区域可见囊性变或坏死。

注:A为HE染色示肿瘤细胞染色质,无明显核仁、核分裂多少不等,×400; B为HE染色示经典的CCSK,见肿瘤细胞巢索被纤细、均匀分布的血管网分隔,部分为透明细胞区,×200; C为免疫组化染色示肿瘤细胞细胞周蛋白D1(CyclinD1)+,×200,EnVision法; D为免疫组化染色示肿瘤细胞波形蛋白(Vimentin)+,×200,EnVision法。图2 肿瘤组织镜下检查
2.3免疫组化染色检查 11例CCSK中阳性表达的标记物有CyclinD1(图2C)、Vimentin(图2D)、肿瘤抑制基因(INI1);阴性表达的标记物有角蛋白(CK)、上皮膜蛋白(EMA)、分化簇34(CD34)、抗肌内膜抗体(Desmin)、抗平滑肌抗体(SMA);其他免疫标记抗体如Bcl-2、CD56、CD99、S100等呈不同程度表达。
3 讨 论
有研究结果显示,CCSK占小儿肾肿瘤的2%~5%,男性发病率稍高于女性,发病率大约为1.3∶1.0,肿瘤常为单侧发病[2],本研究数据结果与以上研究结论不一致,可能与本研究样本较小有关。CCSK患儿常见的临床表现与Wilms瘤相似,常表现为腹部隆起或包块、腹部疼痛及肉眼血尿,影像学检查也无特异性,若影像学检查表现为肾脏肿块内含有较多囊性成分、血供较丰富、伴钙化,特别是发生骨转移征象时,提示CCSK的可能性大[3]。早年研究认为,CCSK是一种易出现骨转移的恶性肿瘤,2005年国际儿科肿瘤学会(SIOP)和2006年美国肾母细胞瘤研究组(NWTSG)的报道中指出,脑已经取代骨,成为CCSK最易转移的部位,其次容易发生转移的部位是肺、后腹腔和肝脏,易发生脑和骨的转移也是CCSK区别于Wilms瘤的关键之处,这也意味着CCSK的预后更差、病死率更高[4-5]。
目前,CCSK的发病机制并不清楚,目前研究尚无家族性或相关综合征的报道,有国外学者研究认为BCOR基因的表达异常与CCSK的发展过程有关[6]。细胞遗传学发现CCSK可以有t(10;17)(q22;p1)和 t(10;17)(q11;p12)染色体异位,少数也可发生14号染色体长臂丢失del(14)(q24.1q31.1),p53基因位于17号染色体,因此,CCSK常伴有p53蛋白的缺失[7]。
CCSK瘤体一般较大,多位于肾中央,可呈分叶状,包膜不明显,切面呈灰白色或淡黄色,实性均质样,伴黏液透明感,可有囊性变及坏死,质地柔韧。经典CCSK镜下组织形态表现为未分化的肿瘤细胞,形态单一,呈条索或团巢状,6~10个细胞群由均匀分布的“鸡爪”样血管网分隔,形成索梁状,细胞核呈卵圆形或略不规则,染色质细腻,核仁不明显,少数核仁有明显核沟。肿瘤细胞因细胞外基质分散,不会拥挤或重叠,呈透明状。该肿瘤组织形态多样,90%表现为经典型,还可表现为上皮样型、梭形细胞型、硬化型、栅栏型、囊肿型、窦腔型、周细胞瘤样型、多形型及间变型[8]。CCSK的免疫组化表型缺乏特异性的免疫标记物,阳性表达的抗体有Vimentin、CyclinD1、INI1,阴性表达的抗体有CK、EMA、CD34、Desmin、SMA。CCSK 起源于原始间胚叶细胞,无上皮分化能力,CK 阴性,Vimentin阳性。CyclinD1在CCSK中的临床价值是近年研究的热点,CyclinD1在鉴别CCSK和富含胚胎型WT方面有很大的帮助[9]。有国外学者研究认为BCOR基因的表达异常与CCSK 的发展过程有关[6,10],组织中BCOR基因或者抗体将可能成为一种特异性更高的肿瘤标记物。
CCSK发病率低,在影像学上缺乏特异性,临床表现也与Wilms瘤很相近,术前很难与Wilms瘤等其他肿瘤鉴别,确诊仍依赖于病理诊断。CCSK需要与Wilms瘤、中胚叶肾瘤及肾横纹肌样瘤(RTK)进行鉴别诊断。(1)Wilms瘤:组织学中Wilms瘤由未分化肾胚芽组织、间叶组织和上皮成分构成,可见原始肾小球、肾小管成分等,上皮成分呈菊形团样。免疫组化标记显示不仅Vimentin、CK为阳性,胚芽成分及上皮成分中WT1也为阳性,第11号染色体异常;而CCSK无原始胚芽成分及上皮成分,Vimentin和Bcl-2阳性,且CK不表达。(2)中胚叶肾瘤:组织学上主要由具有成纤维细胞、肌纤维母细胞及平滑肌细胞特性的梭形细胞构成,排列呈车辐状,无明显透明细胞分化,免疫组化染色显示Vimentin和HHF35阳性,Desmin不同程度表达,遗传学上可见11号染色体三倍体,而CCSK不表达HHF35及Desmin。(3)RTK:RTK是发生于低龄儿童的高侵袭性恶性肿瘤,RTK典型的组织学表现为细胞核大,细胞质内可见嗜酸性包涵体,可见明显的大核仁,免疫组化染色常见Vimentin强阳性,CK和EMA灶性强阳性,另外,RTK细胞核存在特异性INI1表达缺失。而CCSK免疫组化染色不表达CK和EMA 等上皮性标记,INI1阳性。
CCSK的治疗上以根治性肾切除术联合加强化疗方案为主,联合治疗被认为是治疗CCSK的一种非常可靠的治疗方法[11]。虽然CCSK病理形态表现及异型性并没有太过于凶险,但CCSK具有高度侵袭性和广泛转移能力,预后差,其预后与发病年龄、肿瘤分期及坏死程度相关,复发率和病死率也较高。
猜你喜欢
浙江医学(2020年9期)2020-07-01
浙江中西医结合杂志(2019年4期)2019-05-05
浙江医学(2019年2期)2019-01-23
中国市场(2017年5期)2017-03-15
华东师范大学学报(自然科学版)(2017年1期)2017-02-27
高师理科学刊(2016年8期)2016-06-15
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国继续医学教育(2015年1期)2016-01-06
中国癌症杂志(2015年4期)2015-12-09
中国医疗美容(2015年1期)2015-07-12
