
火焰原子吸收光谱法测定食品中钠的不确定度评定
2021-02-15 06:22李煜,王娜,郭杨,陈爽
食品安全导刊 2021年33期
李 煜,王 娜,郭 杨,陈 爽
(无限极(营口)有限公司,辽宁营口 115000)
钠普遍存在于各种食品中,在人体中是一种重要的无机元素,对食品中的钠进行准确定量及可靠性评价具有重要的意义。不确定度是评价结果可靠性的常用度量参数之一,不确定度就是表征合理地赋予被测量之值的分散程度[1]。
本文依据国标GB 5009.91—2017[2],选择火焰原子吸收光谱法对食品中的钠进行测定,参考CNASGL006:2019 化学分析中不确定度的评估指南[1]和JJF 1059.1—2012 测量不确定度评定与表示[3]对火焰原子吸收光谱法测定食品中钠的不确定度进行评定。
1 材料与方法
1.1 材料
本试验中选择的食品为一种植物饮料。
1.2 主要仪器与试剂
PinAAcle 900H原子吸收光谱仪,具有火焰原子化器及钠空心阴极灯(PerkinElmer公司);SQP分析天平(赛多利斯科学仪器(北京)有限公司);DS-360石墨消解仪(中国广州分析测试中心)。
硝酸(优级纯,Merck公司);高氯酸(优级纯,Sigma-Aldrich公司);氯化铯(光谱纯,Aladdin公司);钠标准液(1 000 µg/mL,国家钢铁材料测试中心钢铁研究总院,批号:20031235)。
1.3 试验方法
1.3.1 仪器条件
波长:589.00 nm;狭缝:0.2 nm;灯电流:8 mA;空气流量:10.00 L/min;乙炔流量:2.82 L/min。
1.3.2 标准溶液的配制
(1)标准储备液。准确吸取钠标准液5.0 mL于50 mL容量瓶中,用水稀释至刻度,混匀。该标准储备液的浓度为100 µg/mL。
(2)标准系列使用液。分别准确吸取钠标准储备液0.0 mL、0.5 mL、1.0 mL、2.0 mL、3.0 mL和4.0 mL于100 mL容量瓶中,加氯化铯溶液4 mL,用水定容至刻度,混匀。该标准系列浓度分别为0.0 mg/L、0.5 mg/L、1.0 mg/L、2.0 mg/L、3.0 mg/L和4.0 mg/L。
1.3.3 试样消解
准确称取固体试样0.2~3.0 g(精确至0.001 g)或准确移取液体试样0.500~5.000 mL于消解管中,加入8 mL硝酸、0.5 mL高氯酸,加塞浸泡过夜,放在石墨消解仪中,设置消解程序进行消解。若消化液呈棕黑色,再加少量硝酸,直至冒白烟,消化液呈无色透明或略带黄色,冷却,后用水定容至25~100 mL,混匀备用。同时做空白试验。
1.3.4 试样测定
根据试样溶液中被测元素的含量,需要时将试样溶液用水稀释至适当浓度,并在空白溶液和试样最终测定液中加入一定量的氯化铯溶液,使氯化铯浓度达到0.2%。分别吸取试剂空白液和试样消化液导入调至最佳条件的火焰原子化器中,测定其吸光度值。
2 结果与分析
2.1 数学模型
其中:X-试样中钠的含量,单位为mg/100 g或mg/100 mL;C1-供试液中钠的浓度,单位为mg/L;C0-试剂空白中钠的浓度,单位为mg/L;V-供试液体积,单位为mL;m-试样称样量或移取体积,单位为g或mL。
2.2 测量不确定度来源分析
将钠含量计算数学模型中的C、V、m3个参数作为主要分支,根据试验过程的每个步骤做出因果图,见图1,将图1中类似影响因素组合后做出类似影响因素组合图,见图2,将图2中精密度和正确度表示为重复性和回收率后做出不确定度来源图,见图3。由图3可知,钠含量测定的不确定度来源有:①试样称量引入的不确定度(m);②供试液定容引入的不确定度(V);③供试液中钠浓度引入的不确定度(C);④重复性引入的不确定度(Rep);⑤回收率引入的不确定度(Rec)。

图1 不确定度来源首次列表
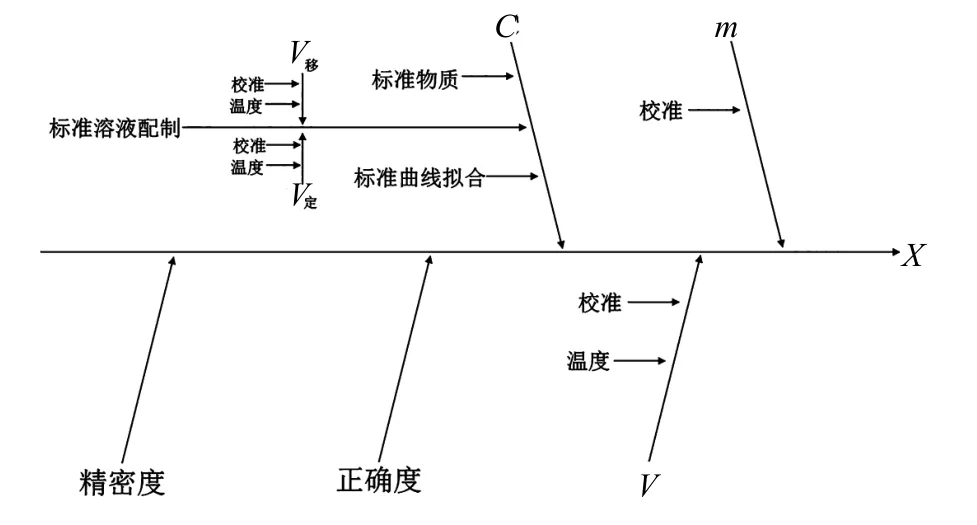
图2 类似影响因素组合
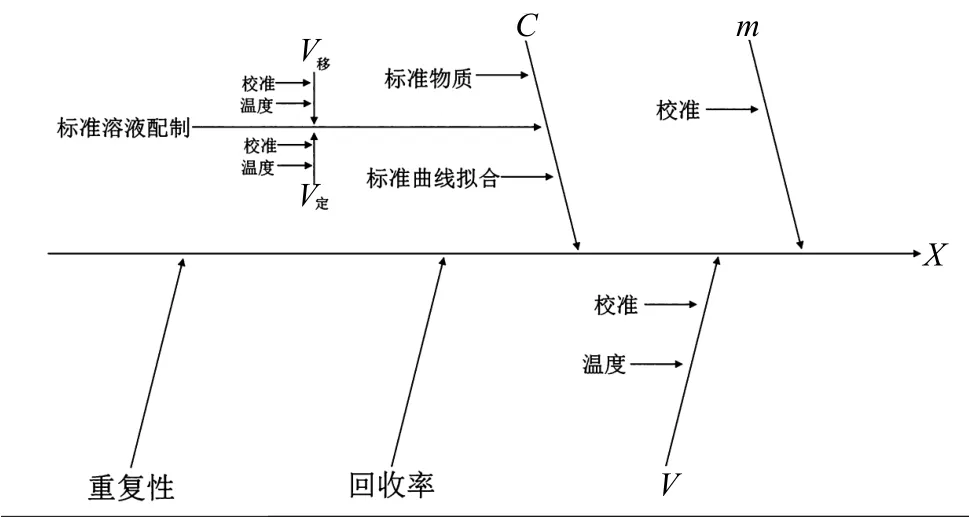
图3 不确定来源图
2.3 钠含量测定的不确定度传播规律
钠含量测定的不确定度传播规律为:
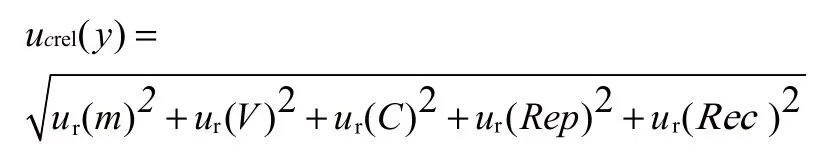
2.4 不确定度分量的量化
2.4.1 试样称量引入的不确定度
准确移取液体试样0.500 mL。使用1 000 µL移液枪,计量证书标明移液500 µL时,容量误差是±0.5 µL,取矩形分布,引入的标准不确定度为相对标准不确定度为
2.4.2 供试液定容引入的不确定度
样品消化后转入100 mL容量瓶中,用超纯水定容至刻度。
(1)JJG 196—2006[4]规定,20 ℃时100 mL A级容量瓶容量允差为±0.10 mL,取三角形分布,引入的标准不确定度为
(2)实际温度与校准时温度不一致,温度变化ΔT为 2 ℃, 水 的 膨 胀 系 数α=2.08×10-4/℃,取矩形分布,则液体膨胀产生的标准不确定度为
2.4.3 供试液中钠的浓度引入的不确定度
供试液中钠的浓度引入的不确定度ur(C)来源有3部分:钠标准液的不确定度、标准液配制引入的不确定度以及标准曲线拟合引入的不确定度。
(1)钠标准液的标准不确定度。钠标准溶液浓度为1 000 µg/mL,标准证书给出的扩展不确定度为U=4 µg/mL,k=2,则标准不确定度为钠标准液的相对标准不确定度为
(2)标准液配制引入的不确定度。单标线吸量管移取5.0 mL标准溶液于50 mL容量瓶中,用超纯水定容至刻度,即得浓度为100 µg/mL的钠标准储备液。分别吸取钠标准储备液0.5 mL、1.0 mL、2.0 mL、3.0 mL和4.0 mL于100 mL容量瓶中,用超纯水定容至刻度。
(a)标准储备液配制时5 mL单标线吸量管 引 入 的 不 确 定 度。JJG 196—2006[4]规 定,20 ℃时,5 mL A级单标线吸量管的容量允差为±0.010 mL,取三角形分布,则标准不确定度为实际温度与校准时温度不一致,室温温度变化ΔT为2 ℃,水的 膨 胀 系 数α=2.08×10-4/℃, 取 矩 形 分 布,液体膨胀产生的标准不确定度为引入的标准不确定度为相对标准不确定度为
(b)标准储备液配制时50 mL容量瓶引入的标准不确定度。JJG 196—2006[4]规定,20 ℃时50 mL A级容量瓶的容量允差为±0.05 mL,取矩形分布,则标准不确定度为实际温度与校准时温度不一致,温度变化ΔT为 2 ℃, 水 的 膨 胀 系 数 α=2.08×10-4/℃, 取矩形分布,液体膨胀产生的标准不确定度为引入的标准不确定度为u(V50mLT)=0.012 009 mL。相对标准不确定度为0.000 474。所以,标准储备液引入的相对标准不确定度为
(c)标准系列使用液配制时5 mL分度吸量管引入的不确定度。JJG 196—2006[4]规定,20 ℃时5 mL流出式A级分度吸量管的容量允差为±0.025 mL,取三角形分布,则吸取5次的标准不确定度为
实际温度与校准时温度不一致,室温温度变化ΔT为2 ℃,水的膨胀系数α=2.08×10-4/℃,取矩形分布,则吸取5次液体膨胀产生的标准不确定度为引入的标准不确定度为0.022 979 mL。相对标准不确定度为ur(V5')=
(d)标准系列使用液配制时100 mL容量瓶引入的不确定度。按照JJG 196—2006[4]规定,20 ℃时100 mL A级容量瓶的容量允差为±0.10mL,取矩形分布,则定容5次的标准不确定度为u(V100mL)=。实际温度与校准时温度不一致,温度变化ΔT为2 ℃,水的膨胀系数α=2.08×10-4/℃, 取 矩 形 分 布, 则 定 容 5次液体膨胀产生的标准不确定度为(V100mLT)=引入的标准不确定度为0.105 913 mL。相对标准不确定度为所以,标准系列使用液引入的相对标准不确定度为0.004 716。综上所述,标准液配制过程引入的相对标准不确定度为
(3)标准曲线拟合时产生的不确定度。吸取钠标准系列使用液,各浓度系列进行3次重复性测定,测其信号值并取平均,结果见表1。
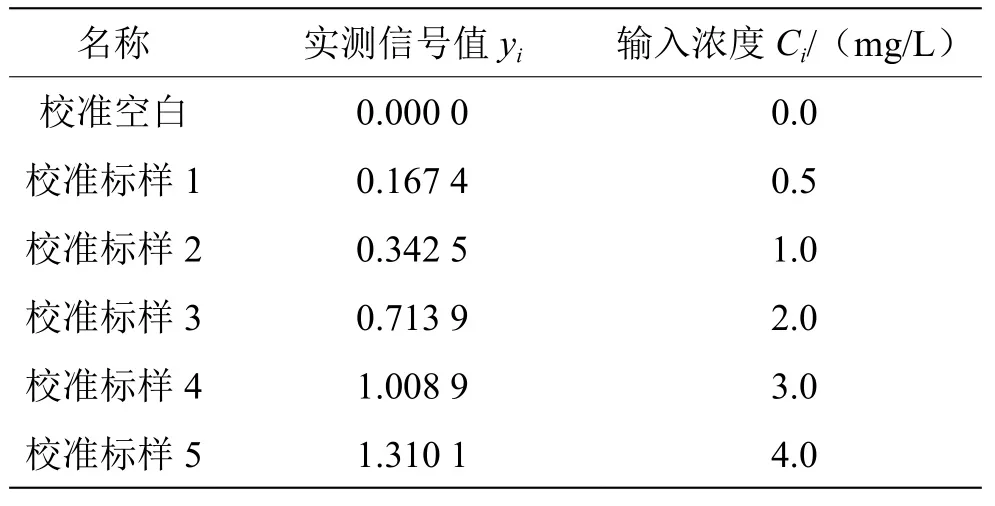
表1 标准系列使用液测定结果
仪器将实测信号值和输入浓度用最小二乘法进行拟合,得到标准曲线方程为y=aC+b=0.330 21x+0.012 61,r=0.999 032。
Ci为输入浓度,c为输入浓度平均值1.75 mg/L,yi为实测信号值,yj为将输入浓度代入曲线得到的校准信号值。残差平方运算结果见表2。

表2 残差平方运算结果
试验得到的供试液中钠浓度C1=0.745 mg/L,则标准曲线拟合时产生的不确定度为
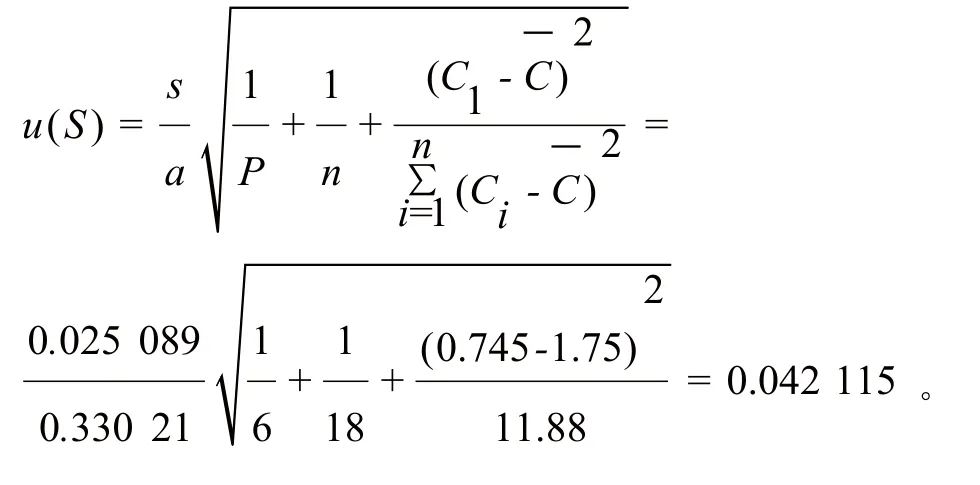
其中:s-标准溶液信号值残差的标准偏差;a-标准曲线的斜率;P-供试液钠浓度的测量总次数,本次评定为6;n-标准溶液的测量总次数,本次评定为18;
C1-供试液中钠浓度值;C-标准曲线浓度平均值。
相对标准不确定度为
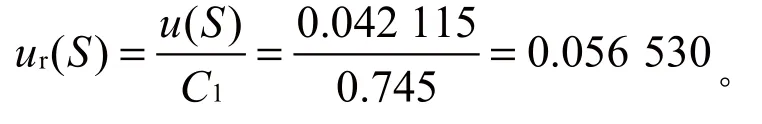
综上所述,供试液中钠的浓度引入的不确定度ur(C)为
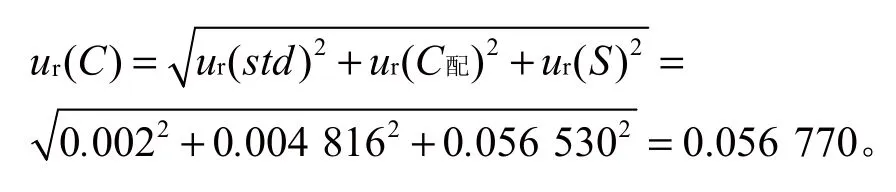
2.4.4 重复性引入的不确定度
相同操作者、相同测量系统、相同操作条件、相同地点,在短时间内对试样进行6次独立测试,结果见表3。
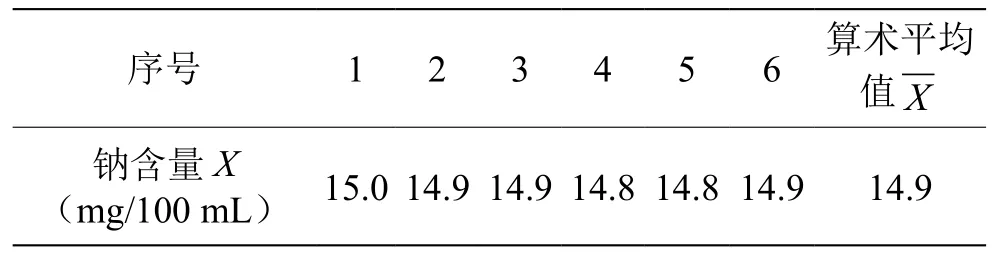
表3 重复性结果
由表3可知,单次测量的标准不确定度为
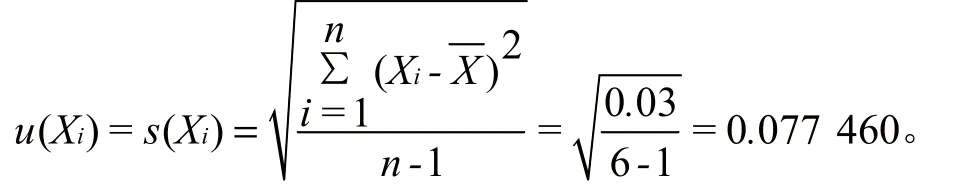
标准不确定度为:

相对标准不确定度为:

2.4.5 回收率引入的不确定度
对加标试样进行6次独立测试,根据实测值和加标量计算得到的回收率结果见表4。

表4 回收率结果
计算6个回收率的标准偏差为s=4.138 997%。
标准不确定度为:
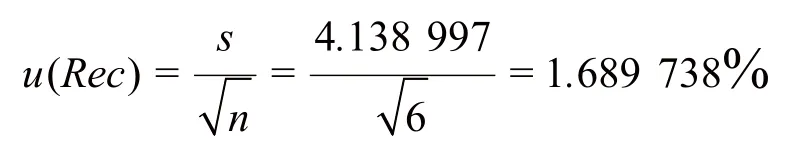
相对标准不确定度为:

通过t检验确认结果是否需要回收率修正:
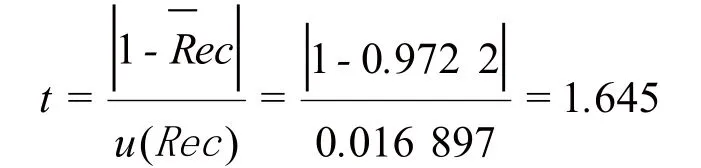
查t检验临界值表[5]得,n-1自由度、95%置信度双侧t(表)=2.571,则t<t(表),无显著性差异,结果不需要用回收率修正。
2.5 合成各相对标准不确定度分量
各相对标准不确定度分量值见表5。

表5 相对标准不确定度分量值
由表5可知,合成相对标准不确定度为:

合成标准不确定度为:
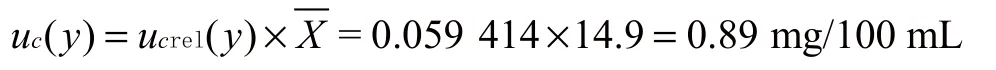
2.6 扩展不确定度的计算
取95%的置信水平,包含因子k=2,则扩展不确定度。
2.7 测量结果及不确定度的表述
本试验食品中钠含量为(14.9±1.8)mg/100 mL,k=2。
3 结论与讨论
由各不确定度分量值可知,测量不确定度主要由供试液中钠浓度引入,其中标准曲线拟合时产生的不确定度最大,所以仪器状态是试验的控制关键点。日常操作过程中,需要对仪器的状态重点关注,采用定期维护、延长预热时间、增加测量次数来确保检测结果的可靠性。
猜你喜欢
中国测试(2022年4期)2022-05-10
中学生数理化·自主招生(2022年4期)2022-05-09
中学化学(2019年4期)2019-08-06
Cancer Biology & Medicine(2016年4期)2017-01-13
电源技术(2015年7期)2015-08-22
电测与仪表(2015年7期)2015-04-09
护理研究(2014年26期)2014-08-15
电测与仪表(2014年9期)2014-04-15
电测与仪表(2014年11期)2014-04-04
化学分析计量(2013年5期)2013-04-10
